










Introduction
Meat is the food containing high-quality protein and contains adequate amounts of essential amino acids and various minerals (Lee et al., 2019). Global per capita meat consumption (kg/person/year) increased from 23.1 kg to 42.2 kg between 1961 and 2011 (Sans and Combris, 2015). Over the centuries, the global goat populations have increased at a much faster rate than the global human population growth rate, primarily in Asia and Africa (Monteiro et al., 2017). Goat meat has been one of the most valuable food resources (Hwang et al., 2019). Goat meat has a similar protein content to chicken, beef, pork, and lamb; however, it is lower in calories, total fat, saturated fat, and cholesterol (Anaeto et al., 2010; Ivanović et al., 2016). In addition, the proportion of unsaturated fatty acids is greater than that of saturated fatty acids (Anaeto et al., 2010).
According to Malekian et al. (2014), Mazhangara et al. (2019), and Shija et al. (2013), eating goat meat may lower the risk of metabolic diseases, such as insulin resistance, type 2 diabetes, cardiovascular diseases, and metabolic syndromes, while also improving the health of a malnourished population. However, the nutritional functionality of goat meat has only been introduced in classic medical literature (Shija et al., 2013), and there is no clear scientific proof. In addition, there are lack of studies to analyze functional components and evaluate their efficacy (Hong et al., 2021).
Therefore, this study evaluated the effects of extracts from the meat of goat forelegs, hind legs, loin, and ribs on α-glucosidase inhibitory activity and the expression levels of the apoptosis-related proteins [Bcl-2-associated X (BAX), p53, and p21] in AGS cells and HT-29 cells. Further, the expression of muscle atrophy proteins [Atrogin-1, muscle atrophy F-box (MAFbx), muscle RING-finger protein-1 (MuRF-1), and myosin heavy chain-7 (MYH-7)] were evaluated in C2C12 myoblasts.
Materials and Methods
To determine the potential health benefits of goat meat, hydrous extract (HE), hot water extract (HWE), and ethanol extract (EE) were prepared from the meat of forelegs, hind legs, loin, and ribs of 12-month-old female goats. Briefly, goat meat (3 g) was pummeled in 15 mL of distilled water by a stomacher (Interscience, Saint-Nom-la-Bretêche, France) for 2 min. The homogenate was then centrifuged at 3,000×g and 25°C for 10 min, and the supernatant was filtered through a No. 1 filter paper (Whatman, Maidstone, UK) to obtain HE. For HWE, 80 mL distilled water was mixed with 20 g ground goat meat in a mixer (Hanil Electric, Wonju, Korea) and heated in a water bath at 100°C for 160 min (Yoo et al., 1990). Subsequently, the mixture of distilled water and goat meat was transferred to a 50-mL conical tube and centrifuged at 1,400×g and 25°C for 30 min. The supernatant was filtered through a No. 1 filter paper to obtain HWE. Ground goat meat (10 g) in 400 mL of 80% ethanol was pummeled by the stomacher for 2 min. The homogenate was left at 25°C for 24 h, transferred to a 50-mL conical tube, and centrifuged at 1,000×g and 25°C for 10 min (Kim et al., 2017). The supernatant was filtered through a No. 1 filter paper, and the filtrate was concentrated at 90°C for 2 h under reduced pressure in a rotary evaporator (Laborota 4001, Heidolph Instruments, Schwabach, Germany).
To measure the degree to which goat meat extract inhibits the digestion and absorption of monosaccharides, the α-glucosidase enzyme reaction of the extracts was evaluated according to the methods developed by Kim et al. (2021) and Si et al. (2010). Briefly, 50-μL aliquots of HE, HWE, and EE were mixed with 50 μL of α-glucosidase (Sigma-Aldrich, St. Louis, MO, USA) in 50 μL of 200 mM potassium phosphate buffer (pH 6.5; Sigma-Aldrich). The mixtures were incubated at 37°C for 10 min before adding 3 mM p-nitrophenyl α-D-glucopyranoside (Thermo Fisher Scientific, Waltham, MA, USA) as a substrate and continuing the reaction at 37°C for 10 min. The reaction was stopped by adding 750 μL of 0.1 M Na2CO3 and centrifuged at 12,000×g and 4°C for 10 min. The supernatant was transferred to a 96-well microtiter plate, and the absorbance at 405 nm was measured with a microplate spectrophotometer (Epoch, BioTek, Winooski, VT, USA) to measure the p-nitrophenol released from the substrate. α-Glucosidase inhibitory activity was calculated as follows:
where A is the absorbance of sample, and B is the absorbance of control.
AGS and HT-29 cells were seeded in 6-well microtiter plates at 5×105 cells/mL in Dulbecco’s modified eagle medium (DMEM; Welgene, Gyeongsan, Korea) supplemented with 10% fetal bovine serum (FBS; Welgene) and 1% penicillin and cultured at 37°C in 5% CO2 incubator for 24 h. The media were then removed, and the cells were washed twice with Dulbecco’s phosphate-buffered saline (DPBS; Welgene). Monolayer cells were exposed to 10-fold diluted EE before being incubated at 37°C for 3 h under 5% CO2. EE was discarded by pipetting after incubation, and the cells were washed twice with DPBS. RNA was extracted from AGS and HT-29 cells by the QIAzol lysis reagent (Qiagen, Hilden, Germany). AGS and HT-29 cells were treated with 400 μL of QIAzol lysis reagent, and the plate was then placed at 25°C for 5 min. Eighty microliters of chloroform were added to the cell suspension in a microcentrifuge tube, and the mixture was then placed at 25°C for 3 min. The clear supernatant was transferred to a microcentrifuge tube after the supernatant was centrifuged at 12,000×g and 4°C for 15 min. Isopropanol (200 μL) was added to the supernatant and the mixture was placed at 4°C for 10 min. The mixture was centrifuged at 12,000×g and 4°C for 10 min, and the cell pellet was then mixed with 400 μL of 75% ethanol. After the mixture was centrifuged at 7,500×g and 4°C, the supernatant was removed, and the pellet was dried in the air for 5 min. The pellet was dissolved in 20 μL of RNase-free water and placed at 55°C–60°C for 10 min. This RNA extract was used to synthesize complementary deoxyribonucleic acid (cDNA) with a QuantiTect Reverse Transcription Kit (Qiagen). For polymerase chain reaction (PCR), 10 μL of AmfiSure qGreen Q-PCR Master Mix (2×; GenDEPOT, Katy, TX, USA), 4 μL of forward primer (1 μM), 4 μL of reverse primer (1 μM), 1 μL of RNase-free water, and 1 μL of cDNA were placed in a 0.1-mL Strip Tubes (Qiagen), and the mixture was amplified as following cycle; 50°C for 2 min (optional uracil DNA glycosylase pre-treatment), 95°C for 3 min (initial denaturation), 40 cycles of 95°C for 5 s (denaturation), and 60°C for 20 s (combined annealing and extension) in Rotor-Gene Q (Qiagen). The primer sequences used in the PCR reaction are presented in Table 1.
| Cell line | Gene | Primer sequence (5’ → 3’) | References | |
|---|---|---|---|---|
| AGS cells | β-actin | F | CGGGAAATCGTGCGTGAC | Perumalsamy et al., 2018 |
| R | AGCTCTTCTCCAGGGAGGA | |||
| bax | F | TTCTGACGGCAACTTCAACTG | ||
| R | GTTCTGATCAGTTCCGGCA | |||
| p53 | F | CCACCATCCACTACAACTACAT | ||
| R | AGGACAGGCACAAACACG | |||
| p21 | F | CTGGAGACTCTCAGGGTCGAA | ||
| R | GGATTAGGGCTTCCTCTTGGA | |||
| HT-29 cells | gapdh | F | GCCGCATCTTCTTTTGCGTC | Özbolat and Ayna, 2021 |
| R | GACGAACATGGGGGCATCAG | |||
| bax | F | GCGAGTGTCTCAAGCGCATC | ||
| R | TGGAGACAGGGACATCAGTCG | |||
| p53 | F | CGACGGTGACACGCTTCC | ||
| R | TTTCCTGACTCAGAGGGGGC | |||
| p21 | F | CACTCCAAACGCCGGCTGATCTTC | Weglarz et al., 2006 | |
| R | TGTAGAGCGGGCCTTTGAGGCCCTC |
AGS and HT-29 cells were seeded in a 6-well microtiter plate at 5×106 cells/mL in DMEM+10% FBS+1% penicillin and incubated at 37°C in 5% CO2 incubator for 24 h. After the incubation, the cell medium was removed, and the cells were washed with DPBS. The cells were treated with 10-fold diluted EE and then incubated at 37°C for 3 h under 5% CO2. The medium was removed, and the cells were washed with chilled (4°C) DPBS. The 200 μL of RIPA buffer (iNtRON Biotechnology, Seongnam, Korea) was added to each well and placed on ice for 30 min to extract the protein from AGS and HT-29 cells. The cell suspension was centrifuged at 15,814×g and 4°C for 15 min, and the supernatant was transferred to a microcentrifuge tube. A DCTM Protein Assay (Bio-Rad Laboratories, Hercules, CA, USA) was used to determine the concentration of the extracted protein. Western blotting was used to measure protein expression as follows; on a 12% SDS-PAGE, 30 μg of protein was separated at 120 V for 1 h. Proteins were then transferred to a polyvinylidene difluoride (PVDF) membrane (GE Healthcare Life Sciences, Marlborough, MA, USA) at 60 V for 2.5 h. After that, the membranes were treated with 5% skim milk (Sigma-Aldrich) at 20°C for 1 h. In order to perform immunoblotting, primary antibodies [{mouse-anti-BAX (ABM40273, 1:300) from Abbkine Scientific, Hubei, China}, {mouse-anti-p53 (sc-100, 1:200) from Santa Cruz Biotechnology, Dallas, TX, USA}, and {rabbit-anti-p21 (ABP57266, 1:300) from Abcam, Cambridge, UK}] were used. As a reference protein, mouse-anti-β-actin (sc-81178, 1:1,000, Santa Cruz Biotechnology) was used. The secondary antibody was goat anti-rabbit IgG-HRP (SA002-500, 1:5,000, GenDEPOT). The immunoreactive bands were visualised by an LAS-3000 Imager (Fujifilm, Tokyo, Japan), and GelQuant software v.2.7. (DNR Bio Imaging Systems, Jerusalem, Israel) was used to quantify the intensity of the bands.
C2C12 myoblasts were seeded in a 6-well microtiter plate at 7×104 cells/mL with DMEM+10% FBS+1% penicillin and cultured at 37°C for 24 h in 5% CO2 incubator. C2C12 myoblasts were differentiated into myotubes by daily replacement with DMEM supplemented with 2% horse serum (Thermo Fisher Scientific) and 1% penicillin/streptomycin (Thermo Fisher Scientific) for 6 days. After the differentiation, 1 mL of 10-fold diluted HWE was added to C2C12 myotubes. To examine the inhibitory effect of goat meat extract on muscle atrophy, 100 μM dexamethasone (Sigma-Aldrich) and 1 mL HWE were added to the myotube cells and incubated at 37°C for 24 h in 5% CO2 incubator (Jeon et al., 2020). The cells were washed twice with DPBS, and RNA extraction and cDNA synthesis were performed according to the methods described in as above. For the measurement of Atrogin-1 and MHC1b gene expression levels, the PCR conditions were same as those described in as above, and the primer sequences are listed in Table 2.
| Gene | Primer sequence (5’ → 3’) | References | |
|---|---|---|---|
| gapdh | F | TGCTGGTGCTGAGTATGTCG | Sultan et al., 2006 |
| R | CAAGCAGTTGGTGGTACAGG | ||
| Atrogin-1 | F | CTCTGTACCATGCCGTTCCT | Jeon et al., 2020 |
| R | GGCTGCTGAACAGATTCTCC | ||
| MHC1b | F | GAGGAAGAGTGAGCGGCG | |
| R | GCCGCAGTAGGTTCTTCCTGT |
C2C12 cells were seeded in at 5×106 cells/mL 6-well microtiter plate containing DMEM+10% FBS+1% Penicillium and incubated at 37°C for 24 h in 5% CO2 incubator. After the incubation, the medium was removed, and the cells were treated with 1 mL of 10-fold diluted HWE and incubated at 37°C for 24 h in 5% CO2 incubator. Muscle atrophy was induced by treating the cells with 100 μM dexamethasone for 48 h. Subsequently, the medium was discarded, and the cells were washed with 1 mL of chilled (4°C) DPBS by pipetting. Protein extraction and quantification of Atrogin-1, MAFbx, MuRF-1, and MYH-7 expression in C2C12 cells were performed according to the methods described in as above. with the following changes: In order to perform immunoblotting, primary antibodies [rabbit-anti-Atrogin-1 (AP2041, 1:700; ECM Biosciences, Versailles, KY, USA), mouse-anti-MAFbx (sc-166806, 1:300; Santa Cruz Biotechnology), mouse-anti-MuRF-1 (sc-398608, 1:300; Santa Cruz Biotechnology), and mouse-anti-MYH-7 (sc-53089, 1:200; Santa Cruz Biotechnology)] were used. Mouse-anti-β-actin (sc-81178, 1:1,000, Santa Cruz Biotechnology) was used as a reference protein. Goat anti-mouse IgG (WG321764, 1:5,000; Invitrogen, Waltham, MA, USA) was used for the secondary antibody.
This experiment was repeated three times. The data (α-glucosidase inhibitory activity, apoptosis transcriptome, apoptosis protein, muscle atrophy transcriptome, and muscle atrophy protein) from the treatments were analyzed with the general linear procedure of SAS® version 9.4 (SAS Institute, Cary, NC, USA). LS means for each dependent variable among the treatments were compared with a pairwise t-test at α=0.05.
Results and Discussion
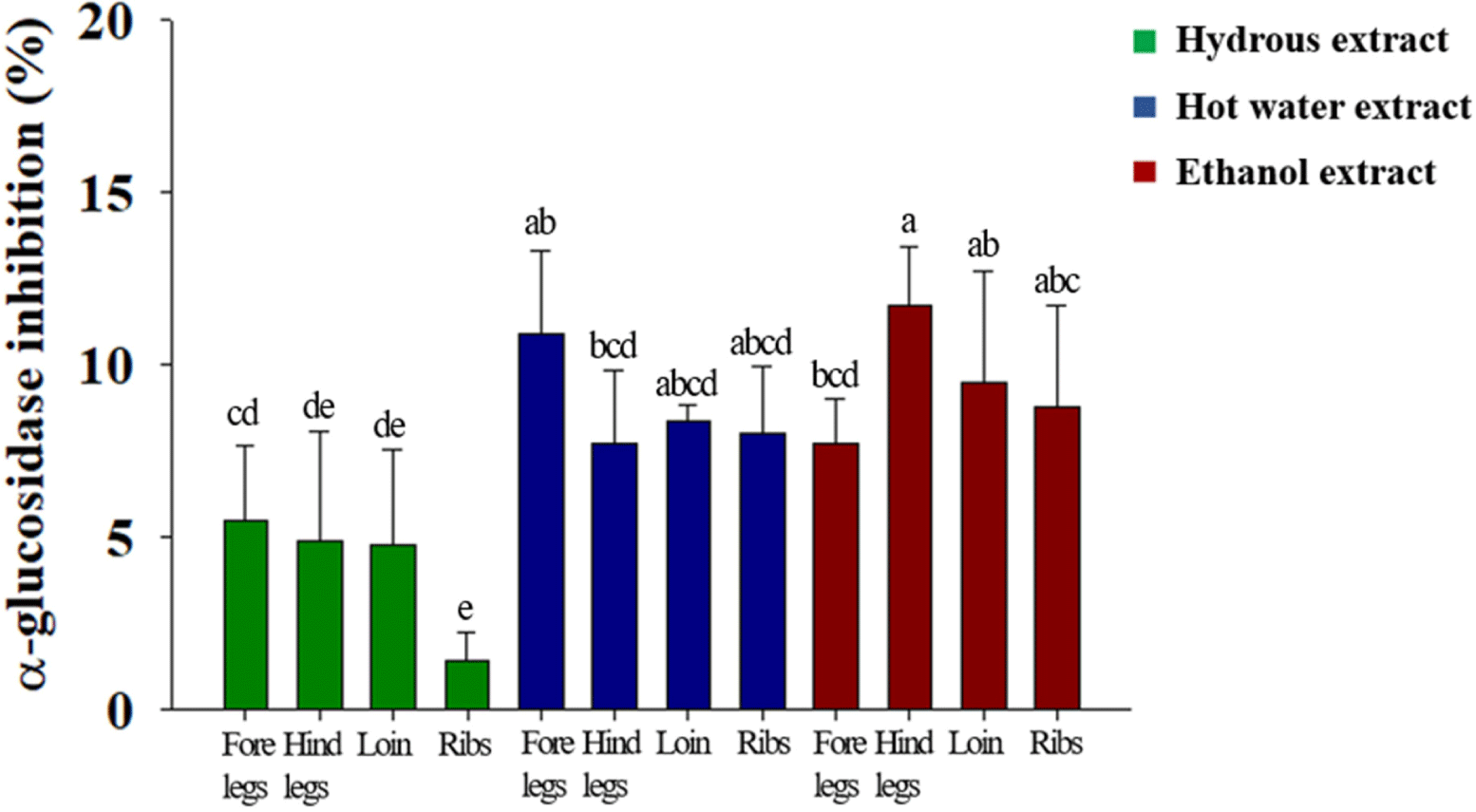
α-Glucosidase is a glycolytic enzyme that is present in the intestinal mucosa. When its enzymatic reaction is inhibited, glucose absorption in the small intestine is delayed, thereby preventing a rapid increase in blood glucose levels after a meal (Robertson et al., 2004). In this study, the α-glucosidase inhibitory activities of HE, HWE, and EE were examined. The α-glucosidase inhibitory activity of EE especially from hind legs was higher (p<0.05) than that of HE and HWE (Fig. 1). HE from ribs had the lowest (p<0.05) α-glucosidase inhibitory activity among 3 goat meat extracts. These results indicate that the fat-soluble component of goat meat may help to prevent a rapid rise in blood sugar.
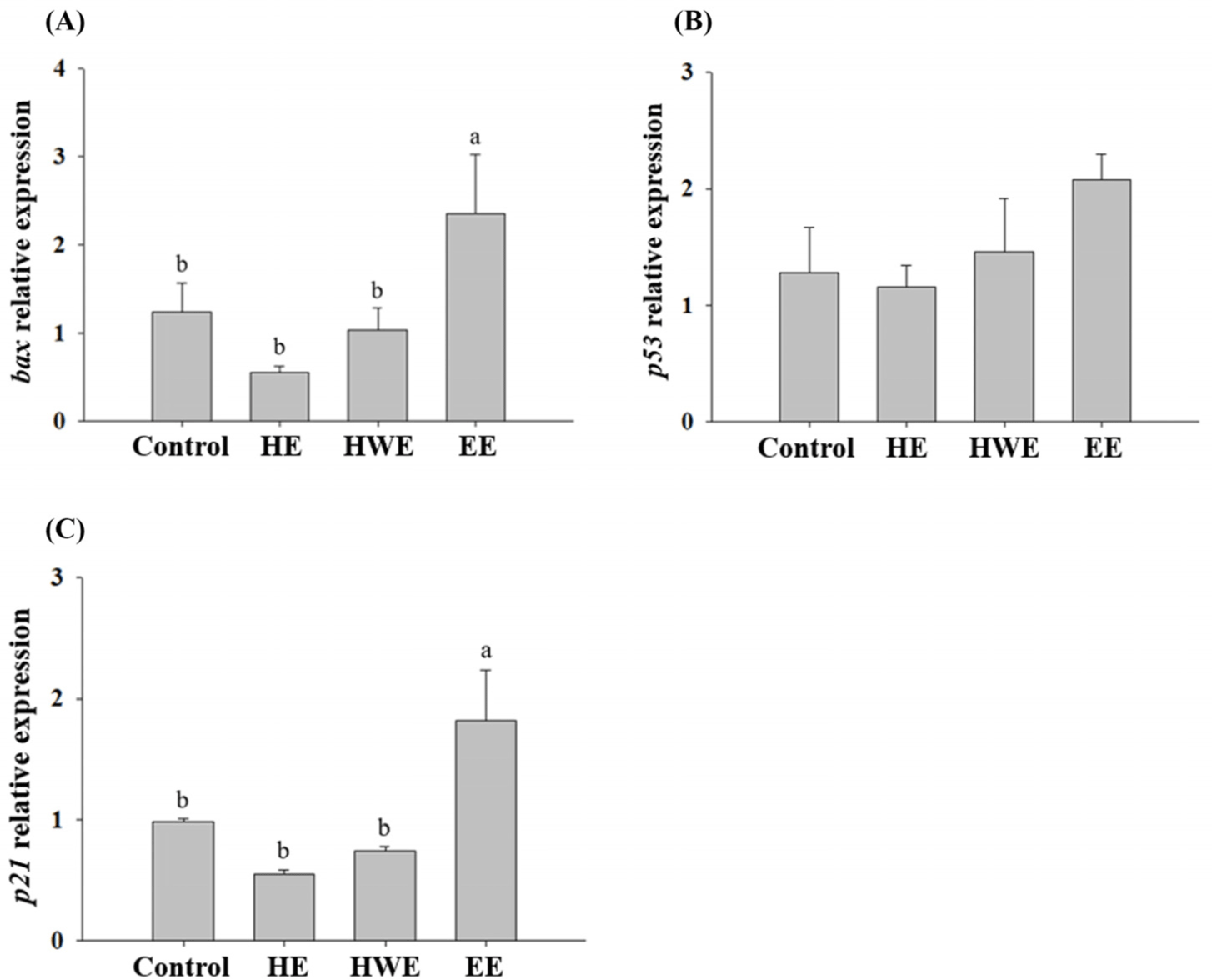
The expression levels of bax, p53, and p21 in HT-29 cells treated with HE, HWE, and EE of goat hind legs meat were examined in a preliminary experiment. Of the extracts, EE caused higher expression levels of bax, p53, and p21 than the other treatments (Fig. 2). Thus, EE was subjected to the further study. Disruption of the apoptotic mechanism is a major cause of normal cells changing into cancer (Hu and Kavanagh, 2003). Apoptosis can be induced by BAX, p53, and p21 (Jäättelä, 2002; Reed, 2002). p21 is strongly induced by p53, as p21 has an important effect on cell cycle arrest. p53 is the best-known tumour suppressor, and its main functions are the induction of apoptosis and cell cycle arrest (Gnanapradeepan et al., 2018). Moreover, it induces p21 to inhibit the growth of malignant tumour cells when DNA abnormalities are detected, and p21 has been shown to mediate G1 cell cycle arrest (El-Deiry et al., 1993; El-Deiry et al., 1994). Galanos et al. (2018) found that upregulated p21 level arrested the AGS cell cycle in vitro. AGS cells treated with EE showed a tendency to increase the expression levels of bax and p53 compared to those of control (cells treated with no EE; Fig. 3). bax, a pro-apoptotic molecule known to induce apoptosis (Yamabe et al., 2014), was expressed in AGS cells at the level similar to those of control when AGS cells were treated with loin EE. BAX expression levels in AGS cells treated with foreleg and loin EE were higher (p<0.05) than those of control (Fig. 4). The expression levels of p53 were higher (p<0.05) in the cells treated with foreleg EE, but not in those treated with hind legs, loin, and rib EE than those of control. The expression levels of p21 tended to be higher in the cells treated with forelegs, hindlegs, loin, and rib EE than in the cells treated with no EE, but this increase was not significant.
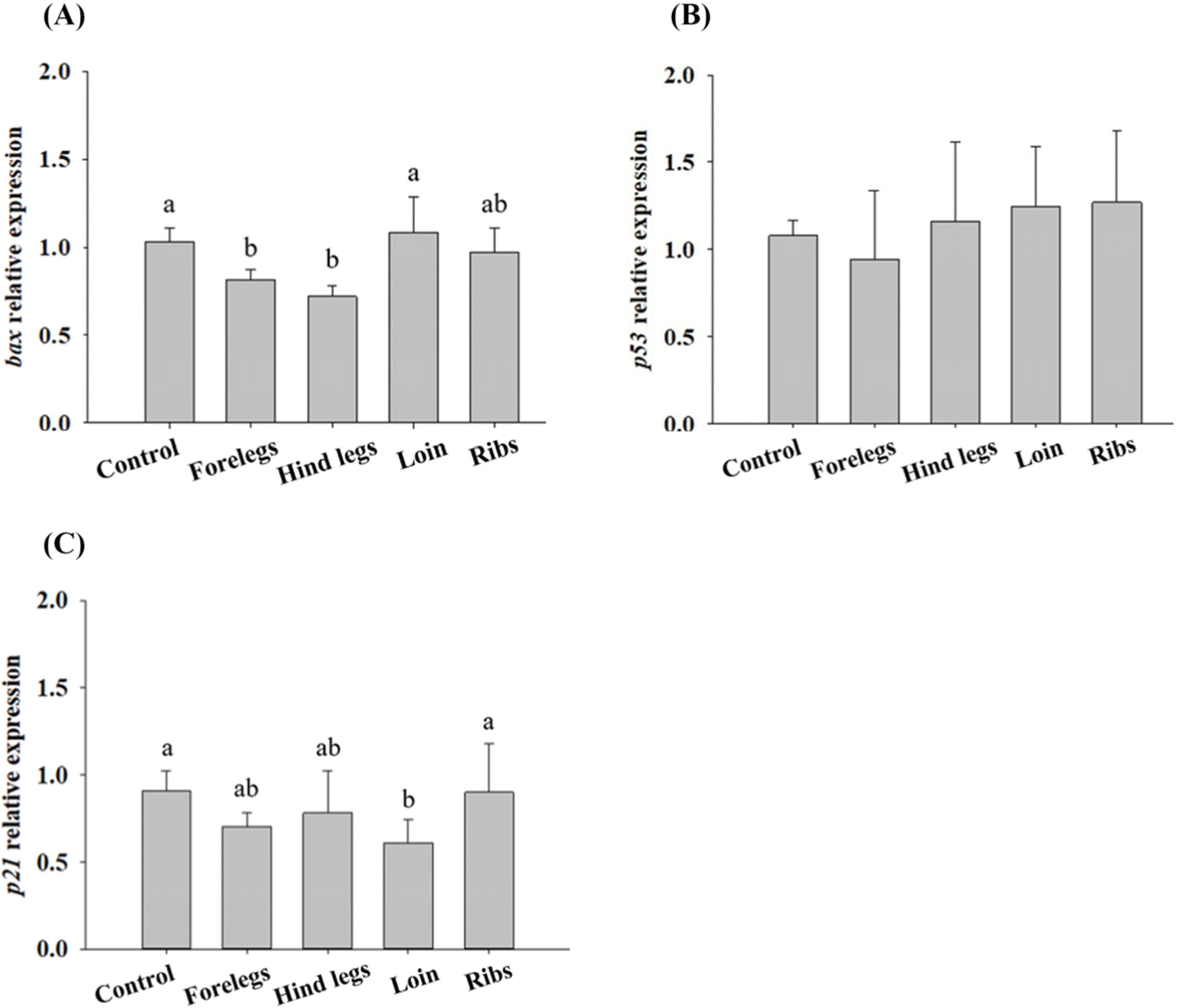
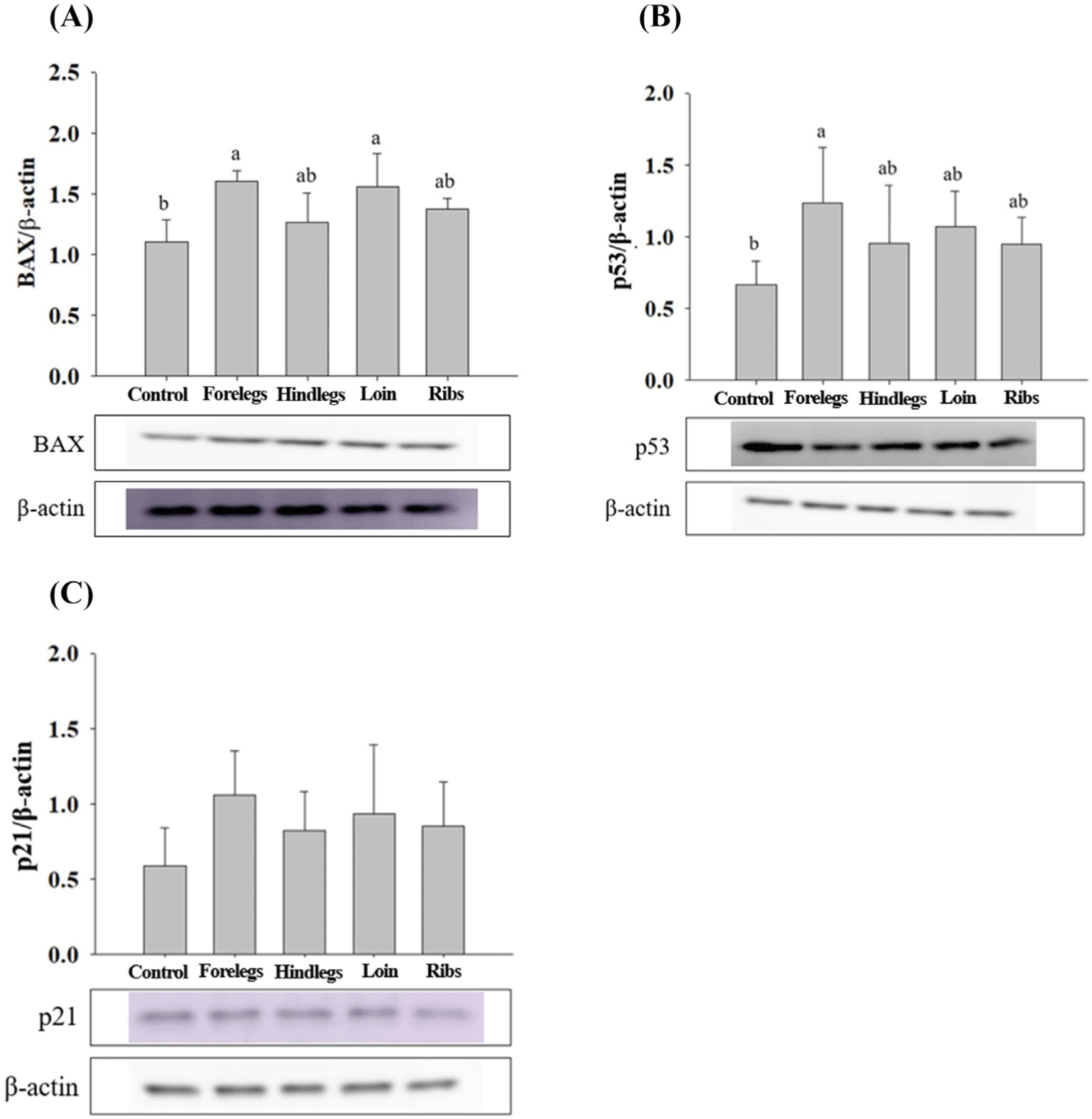
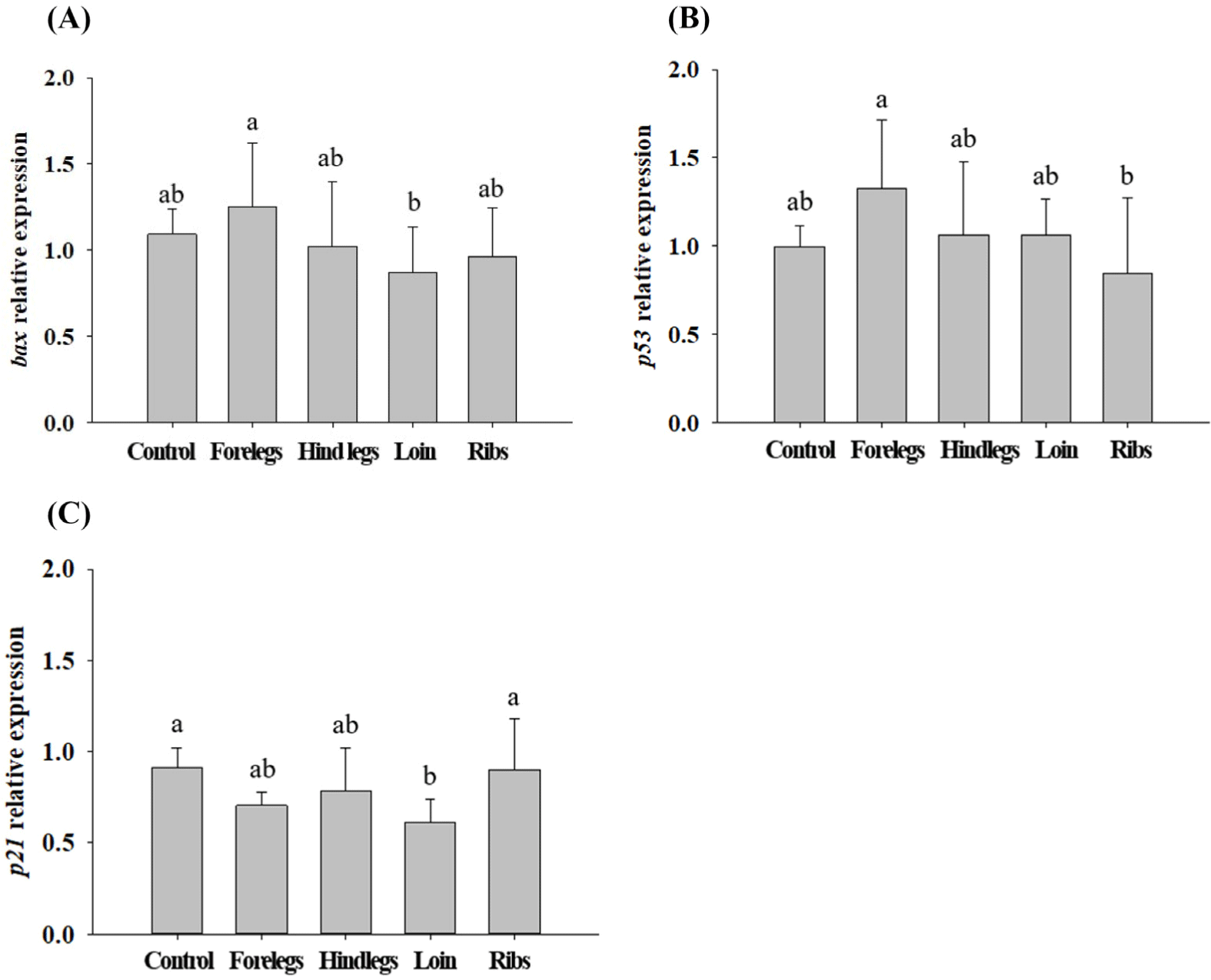
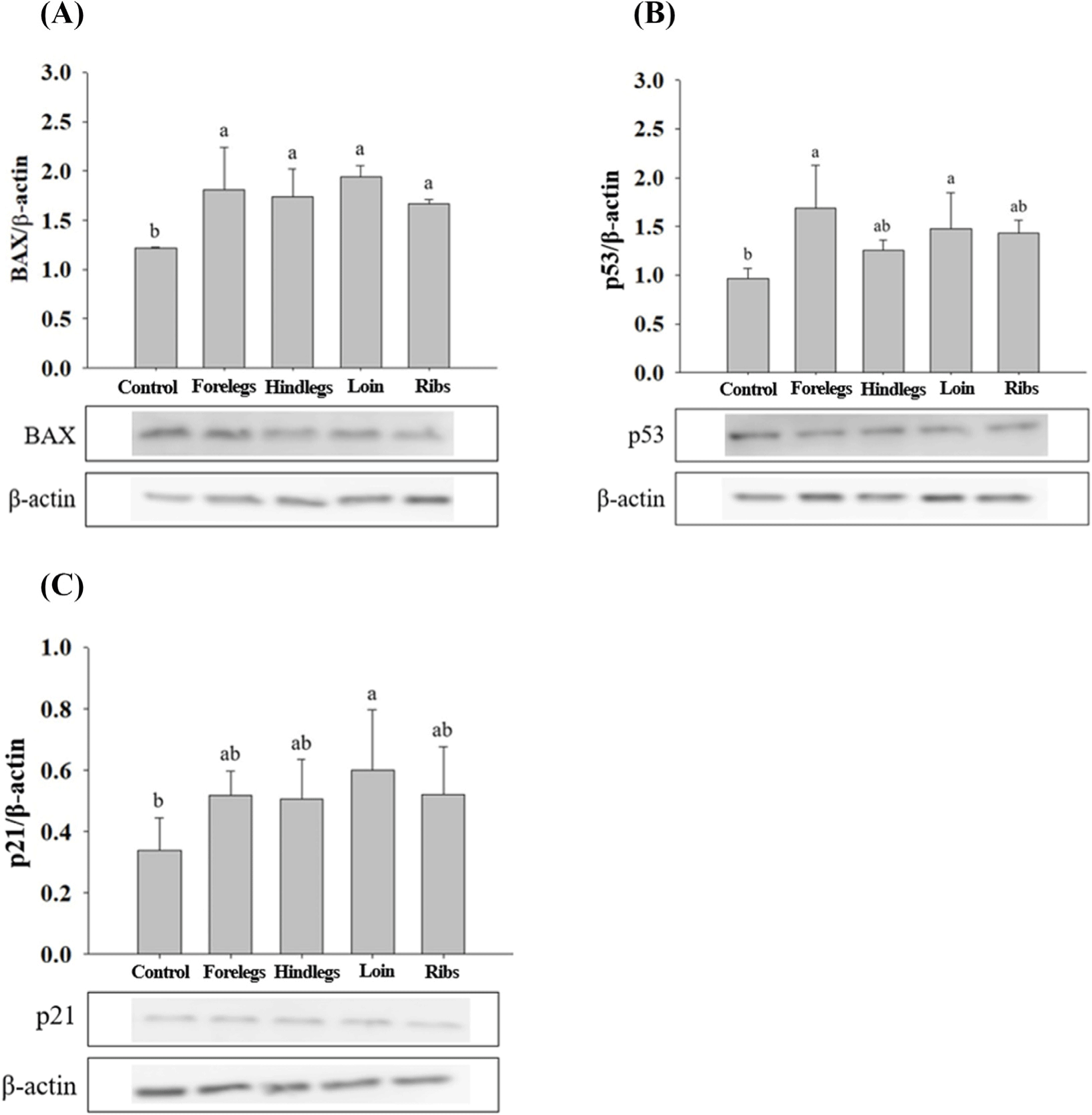
HT-29 cells treated with EE of goat forelegs showed a tendency to increase the expression levels of bax and p53 compared to those of control (cells treated with no EE; Fig. 5). In EE-treated HT-29 cells, especially with the forelegs, hind legs, loins, and ribs extracts, the expression levels of BAX were higher (p<0.05) than those of the control (Fig. 6). p53 expression levels were higher (p<0.05) in cells treated with EE of the forelegs and loin, and p21 expression levels in those treated with loin EE were higher (p<0.05) compared to those of control.
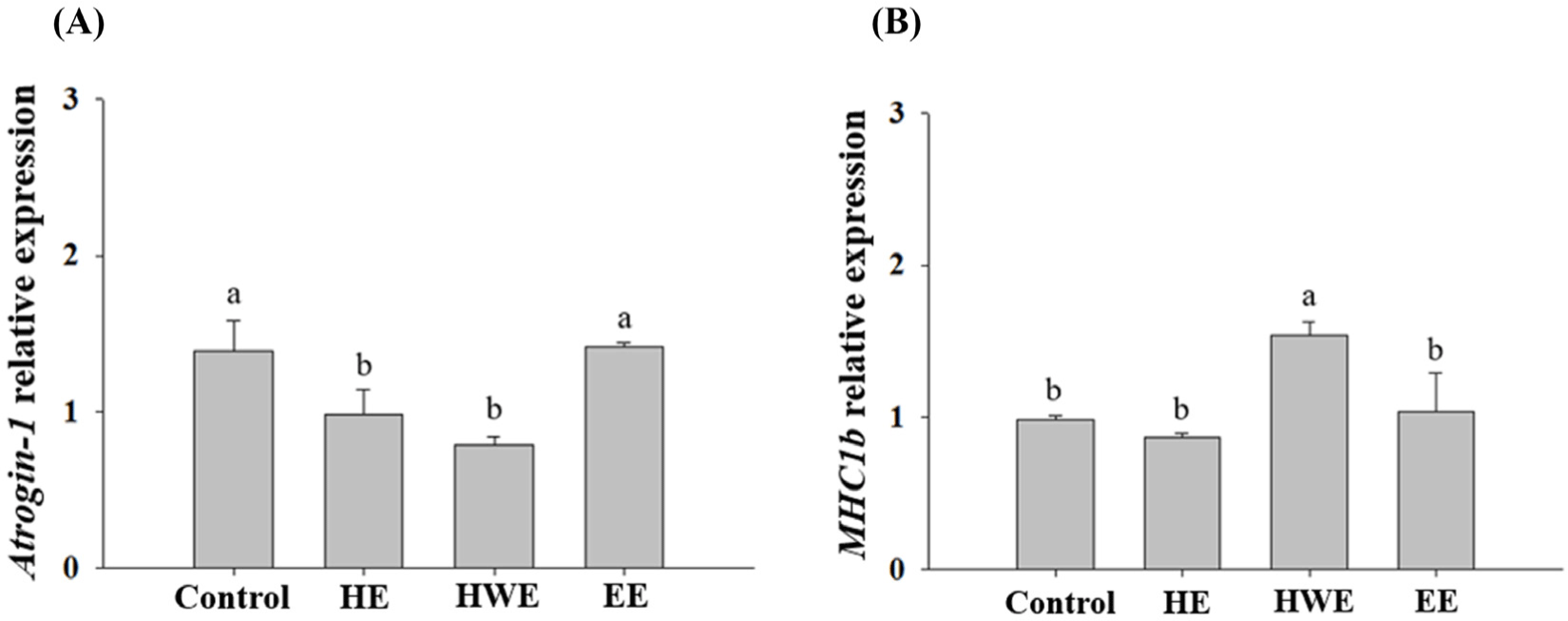
The expression levels of Atrogin-1 and MHC1b in C2C12 cells treated with HE, HWE, and EE of goat hind legs meat were investigated in a preliminary experiment. Of the extracts, HWE caused lower Atrogin-1 expression level and higher MHC1b expression level than the other treatments (Fig. 7). Thus, HWE was subjected to the further study. In C2C12 cells exhibiting muscle atrophy upon treatment with HWE of the forelegs, hind legs, loin, and ribs, the expression of Atrogin-1 which promotes muscle protein degradation (Gumucio and Mendias, 2013) was low (p<0.05; Fig. 8). MHC is abundant in the skeletal muscle, which allows the muscles to contract normally (Rajab et al., 2000). During cell differentiation, genes encoding contractile proteins such as MHC are activated (Brown et al., 2012). A decrease in MHC contributes to decreased muscle mass and contractile function (Balagopal et al., 1997). In our study, an increase in MHC expression was observed. In cells treated with foreleg HWE, MHC1b expression levels tended to be higher than those of the cells with induced muscular atrophy (Fig. 8).
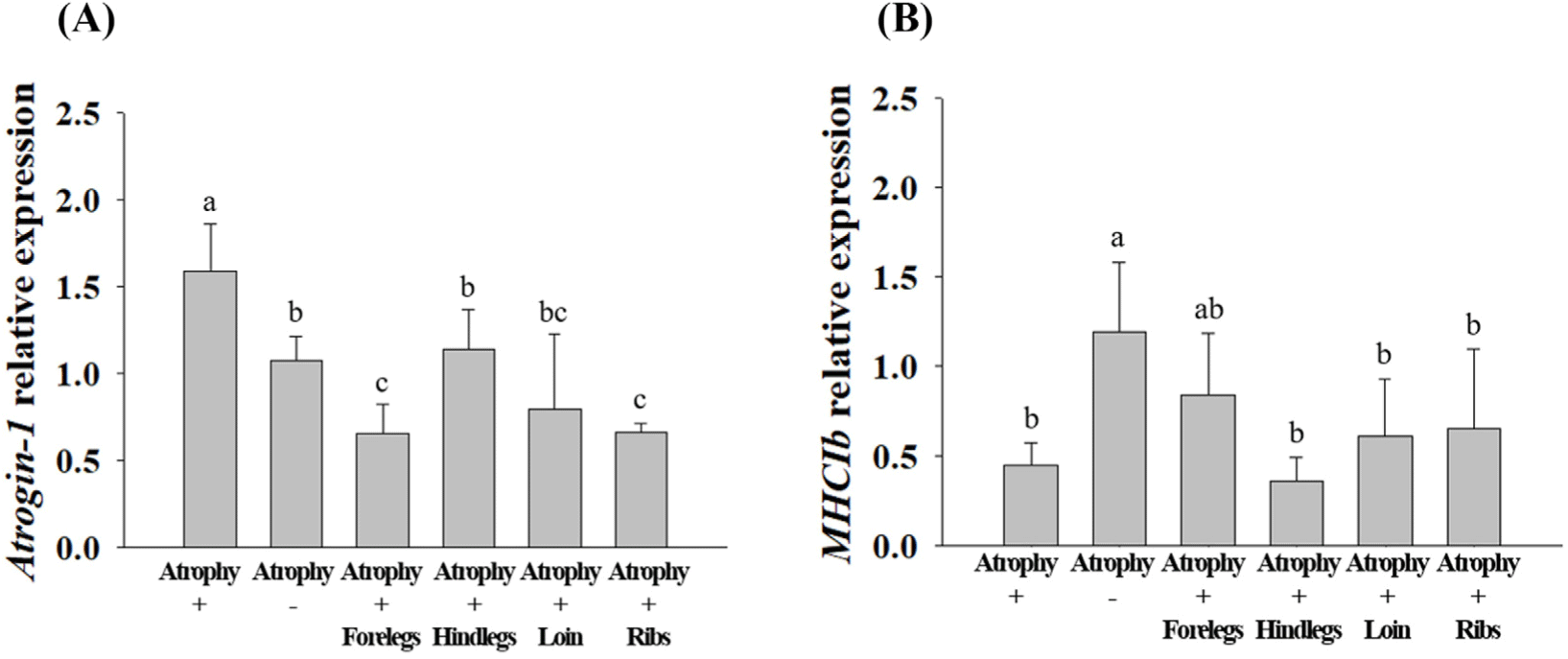
The expression levels of anti-muscle atrophy-related proteins (Atrogin-1, MAFbx, MuRF-1, and MYH-7) in C2C12 cells were analysed (Fig. 9). MAFbx and MuRF-1 are the main skeletal muscle atrophy factors along with Atrogin-1. These factors induce skeletal muscle atrophy in various stressful environments, such as cell aging and glucocorticoid increase (Bodine and Baehr, 2014). The underlying cellular mechanism involves the glucocorticoid receptor and fork head box O (FOXO) transcription factor which increases the expression of MAFbx and MuRF-1, leading to skeletal muscle atrophy (Ebert, 2019; Jo et al., 2021). The MYH-7, encoded by MHC1b, reduces the proportion of muscle fibers with reduced physical activity, such as in chronic disease (Gan et al., 2013; Saltin et al., 1977; Zierath and Hawley, 2004).
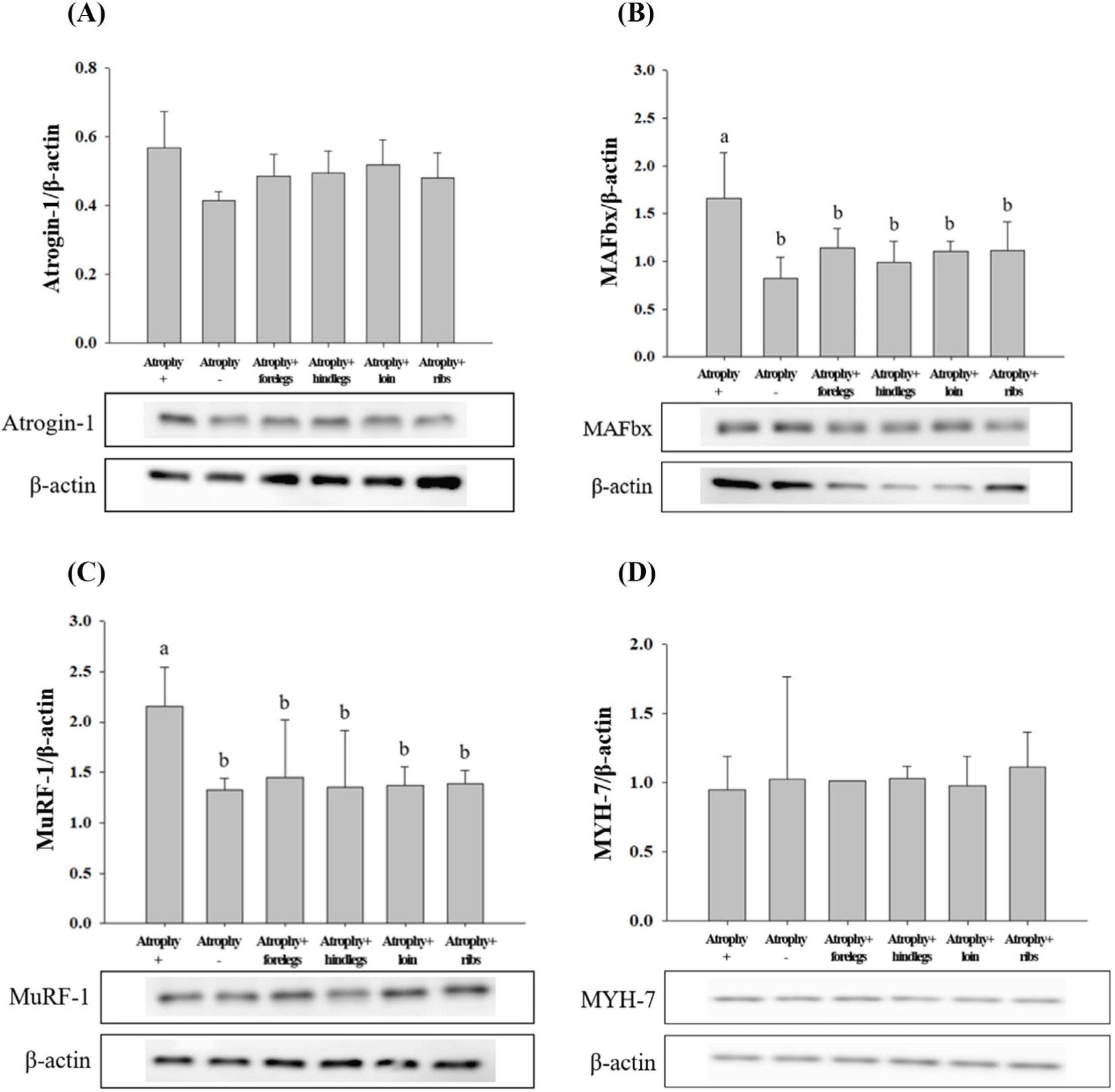
After muscle atrophy was induced, Atrogin-1 expression level was lower (p<0.05) in HWE-treated cells than in atrophy-induced cells; however, there was no significant difference in its expression level among the treatments. In treatments with forelegs, hind legs, loin, and ribs HWE, the expression levels of MAFbx and MuRF-1, which degrade proteins by binding to ubiquitin (Bodine and Baehr, 2014), were similar to those in cells that muscle atrophy was not induced. The expression levels of MAFbx and MuRF-1 were lower (p<0.05) in the cells treated with HWE of the forelegs, hind legs, loin, and ribs than in the muscle atrophy-induced cells. After muscle atrophy was induced, the expression level of MHC1b was higher in the cells treated with foreleg HWE than in the cells with induced muscle atrophy; however, the difference was not significant. Similarly, for MYH-7, there was no significant difference among the different treatment groups.
Goat meat contains more leucine than pork and lamb (Pellett and Young, 1990). Leucine is an essential amino acid that transduces signals during muscle synthesis. The mRNA expression of MAFbx and MuRF-1 were down-regulated by leucine treatment in rats (Wan et al., 2017). Treatment of C2C12 with leucine suppressed the mRNA level of Atrogin-1 (Herningtyas et al., 2008). Haegens et al. (2012) reported that leucine elevated MHC expression and induced myofibril protein adhesion in skeletal muscle. In this study, the expression levels of MAFbx and MuRF-1 in myotubes treated with HWE in cells induced by muscle atrophy were down-regulated (p<0.05) compared to those in cells induced only by muscle atrophy. Thus, it could be considered that leucine in goat meat may help to prevent skeletal muscle atrophy.
Conclusion
This study demonstrated that the in vitro α-glucosidase inhibitory activity of goat meat extracts, and expression levels of BAX, p53 and p21 were increased in goat meat extract-treated AGS and HT-29 cells; in contrast, the expression levels of MAFbx and MuRF-1 in C2C12 myoblasts were decreased. These results were obtained only from in vitro experiments. Thus, the results need to be verified in the experiment of in vivo in a future study.