










Introduction
Non-steroidal anti-inflammatory drugs (NSAIDs) are extensively used as veterinary drugs to prevent inflammation or to treat allergies, fever, and pain. Flunixin is the only NSAID in the U.S. labeled for use in beef and dairy cattle to treat respiratory tract disease and mastitis (Smith et al., 2008). However, it has adverse side effects, such as gastrointestinal bleeding, intestinal ulceration, aplastic anemia, and the inhibition of platelet aggregation (Kari et al., 1995). Daeseleire et al. (2003) proposed a withdrawal time of 12 h owing to the presence of trace residues of 5-hydroxy flunixin after 10 h. According to the U.S. Food and Drug Administration (FDA), the withdrawal times of flunixin are 24 h at a dose of 2.2 mg/kg and 12 h at 1.1 mg/kg for intravenous administration. Moreover, the slaughter and milk withdrawal times are 4 d and 36 h, respectively (FDA, 2004). And tolerance/Safe Level of 5-hydroxy flunixin is 2 μg/kg in milk (FDA, 2020) and the final acceptable daily intake for carcinogenicity was established at 0.72 μg/kg bw/day for human exposure (FDA, 2017).
Flunixin is easily metabolized, and approximately 90% is excreted in the urine (Levionnois et al., 2018). 5-Hydroxy flunixin, one of its metabolites, is a marker in bovine milk, and the residue of flunixin in bovine muscle can be identified in its original form (Chen et al., 2019; Jedziniak et al., 2016; Ngoh et al., 2003). In 2011, the U.S. FDA presented plans to surveil 5-hydroxy flunixin in milk beginning with residue violations of flunixin in dairy cows. This event increased concerns about the presence of drug residues in milk. The European Union (EU), under Community Regulation 2377/90, set the maximum residue level (MRL) for flunixin and its metabolite (5-hydroxy flunixin) at <40 μg/kg in milk produced for human consumption, while the MRL of flunixin is 20 μg/kg in bovine muscle (EU, 1990). In South Korea, flunixin in animal-based foods is regulated by the MRL (MFDS, 2023a). Flunixin residue is defined as its original form, and the MRLs are 0.02, 0.05, 0.01, and 0.02 mg/kg for beef, pork, and horse muscles and milk, respectively. In 2022, the definition was amended for 5-hydroxy flunixin in milk by the Ministry of Food and Drug Safety (MFDS). Therefore, a method for analyzing 5-hydroxy flunixin in milk is required to protect public health.
5-Hydroxy flunixin has a fluorescent property and can be detected at excitation and emission wavelengths of 360 and 495 nm, respectively, using fluorescence detection (FLD; Gallo et al., 2010). Analyzing residues in agricultural products is challenging due to the matrix effects caused by co-extractives, such as pigments, fatty acids, sugars, and other interrupting substances (Fialkov et al., 2007). For the treatment of food samples, a clean-up process is required to remove interfering substances. Solid-phase extraction (SPE) using OASIS HLB, Strata X, and ABN has been used for deproteinization with trichloroacetic acid; however, it causes the loss of certain target compounds of NSAIDs (Malone et al., 2009). Plasma samples have been acidified using inorganic acids, such as hydrochloric acid, to precipitate proteins (Jedziniak et al., 2016). Quick, Easy, Cheap, Effective, Rugged, and Safe (QuEChERS) is the most common method for food sample preparation and is widely used for multi-class, multi-residue analyses (Hajrulai-Musliu et al., 2021; Jang et al., 2022; Wilkowska and Biziuk, 2011). To determine flunixin and 5-hydroxy flunixin contents, high-performance liquid chromatography (HPLC)-ultraviolet spectrometry (Asea et al., 2001; Jedziniak et al., 2016), liquid chromatography (LC)-tandem mass spectrometry (MS/MS; Daeseleire et al., 2003; Kruve et al., 2008; Malone et al., 2009), and other methods have been used. Flunixin is a substance classified and managed as group B (pharmacologically active substances authorised for use in food-producing animals), and the FLD method was suggested in the European Council decision 2002/657/EC (European Commission, 2002). This study attempted to analyze using QuEChERS, which has a simple pretreatment process, and LC-MS/MS, which can analyze various residual animal medicines.
The number of regulated veterinary drugs has continually increased, and the surveillance of residues is becoming stricter with systems such as the positive list system (PLS; MFDS, 2023a). The PLS regulates all veterinary drugs in all types of animal-based foods at a specific concentration (0.01 mg/kg). Therefore, this study aimed to develop a quantitative method for the detection of 5-hydroxy flunixin in milk. By applying 5-hydroxy flunixin, which is not included in the multi-class, multi-residue analysis method established by the MFDS, the applicability was assessed to improve monitoring efficiency and prepare for PLS.
Materials and Methods
Standards (flunixin and 5-hydroxy flunixin) were obtained from Toronto Research Chemicals (TRC; Windsor, CT, USA) and stored at –80°C. HPLC-grade acetonitrile, dichloromethane, n-hexane, methanol, and water were purchased from Merck (Rahway, NJ, USA). The ethylenediaminetetraacetic acid (EDTA) disodium salt solution and formic acid were purchased from Sigma-Aldrich (St. Louis, MO, USA). C18 sorbent was obtained from CHROMAtific (Heidenrod, Germany), and polytetrafluoroethylene (PTFE) and nylon syringe filters were obtained from Teknokroma (Barcelona, Spain). All standard solutions were individually prepared at 1,000 mg/L in methanol and stored at –80°C.
The analysis was performed using a Shimadzu LCMS-8060 system (Shimadzu, Osaka, Japan) connected to a Waters X select HSS C18 column (150 mm×2.1 mm id, 3.5 μm). The mobile phases A and B were 0.1% formic acid in water (A) and 0.1% formic acid in acetonitrile (B), respectively. The LC gradient conditions were as follows: 5% B (0 min), hold at 5% B (0.5 min), increase to 60% B (5.5 min), increase to 100% B (6.0 min), hold at 100% B (10.0 min), rapidly decrease B to 5% (10.2 min), and hold for stabilization (12.00 min). The flow rate, injection volume, and temperature were 0.3 mL/min, 5 μL, and 40°C, respectively. The ESI (electrospray ionization) mode was used, the capillary temperature and voltage were 500°C and 3.6 kV, respectively, and argon gas was used as the impact gas. The specific multiple-reaction monitoring (MRM) conditions are listed in Table 1.
The applicability of the multi-residue, multi-class analysis method developed by the MFDS (2023b) was confirmed as follows. Homogenized samples (2 g) were prepared in a 50-mL centrifuge tube, and 10 mL of water: acetonitrile (1:4, v/v) was added to the sample. The mixture was shaken for 5 min and centrifuged at 4,700×g for 10 min at 4°C. The supernatant was decanted into a second 50-mL centrifuge tube containing 500 mg of C18 and 10 mL of n-hexane saturated with acetonitrile. The mixture was shaken for 10 min and centrifuged at 4,700×g for 5 min at 4°C. Acetonitrile extract (5 mL) was transferred to a 15-mL centrifuge tube and concentrated at 40°C to dry under nitrogen gas. The concentrate was dissolved with 1 mL of water: methanol (1:1, v/v) and filtered through a 0.2-μm PTFE syringe filter. The final sample was analyzed using LC-MS/MS.
The analysis methods developed in Korea by Shin and Choi (2022) for flunixin and 5-hydroxy flunixin were compared with Method 1. Milk samples (2 g) were prepared in a 50-mL centrifuge tube. EDTA solution (100 μL) was added, and the mixture was shaken for 3 min. Subsequently, 8.5 mL of acetonitrile containing 0.1% formic acid and 1.5 mL of dichloromethane were added. The mixture was shaken for 15 min and centrifuged at 4,500×g for 10 min at 4°C. The supernatant was decanted into a 15-mL centrifuge tube containing 150 mg of C18. The mixture was shaken for 10 min and centrifuged at 4,500×g for 10 min at 4°C. The supernatant was transferred to a second 15-mL centrifuge tube and concentrated to dryness under gentle nitrogen gas at 40°C. The concentrate was reconstituted with methanol (1 mL), sonicated for 5 min, and vigorously mixed for 1 min. After centrifuging for 10 min (at 16,000×g and 4°C) and filtering through a 0.2-μm nylon syringe filter, the final sample was analyzed using LC-MS/MS.
According to the Codex guidelines (CAC/GL 71-2009), the method was validated for linearity, accuracy, precision, and detection limits (Codex Alimentarius, 2014). Linearity was represented as the coefficient of determination (r2), and the five points of the calibration curve were at 2.5, 5, 10, 20, and 40 μg/kg. Accuracy and precision were confirmed by repeating the analysis seven times at three level and were represented as the recovery and coefficient of variation (CV), respectively. The detection limits were expressed as limits of detection (LOD) and quantification (LOQ), calculated as 3.3 and 10 times the slope SD, respectively. To confirm inter-lab reproducibility, method validation was performed in two other laboratories.
The matrix effects of the two methods were estimated by calculating the calibration slope (Kwon et al., 2012). Target compounds were spiked into each matrix at six points within the range of 0–80 μg/kg to prepare calibration curves and calculate slope factors. Then, the slope factor was obtained from the calibration curve created by spiking the same amount into the solvent. The matrix effects (%) of the analytes were calculated using the following Eq. (1):
Ferrer et al. (2011) classified the matrix effects as no effect (<20%), medium effect (20%–50%), or strong effect (>50%). Matrix effects were evaluated by referring to the classification.
Results and Discussion
This study presented a quantitative analysis method for 5-hydroxy flunixin residues in milk using flunixin and compared the two methods to increase monitoring efficiency (Tables 2 and 3). Both methods exhibited good linearity (>0.99). In Method 1, the recoveries and CVs were 110%–115% and 2.2%–5.4% for flunixin and 94.0%–108% and 3.1%–9.3% for 5-hydroxy flunixin, respectively. In Method 2, the recoveries and CVs were 97.2%–99.6% and 2.2%–3.9% for flunixin and 84.6%–101% and 0.7%–8.4% for 5-hydroxy flunixin, respectively. The results of both methods met the performance criteria set by Codex for quantitative analytical methods for veterinary drug residues in foods. Therefore, Methods 1 and 2 were suitable for identifying and quantifying flunixin residues in milk products.
| Target | Linearity (r2) | Fortification (μg/kg) | Recovery (%) | Intra-lab CV (%) |
|---|---|---|---|---|
| Flunixin | 0.9991 | 10 | 99.6 | 3.0 |
| 20 | 99.1 | 3.9 | ||
| 40 | 97.2 | 2.2 | ||
| 5-Hydroxy flunixin | 0.9993 | 10 | 101 | 4.1 |
| 20 | 93.6 | 0.7 | ||
| 40 | 84.6 | 8.4 |
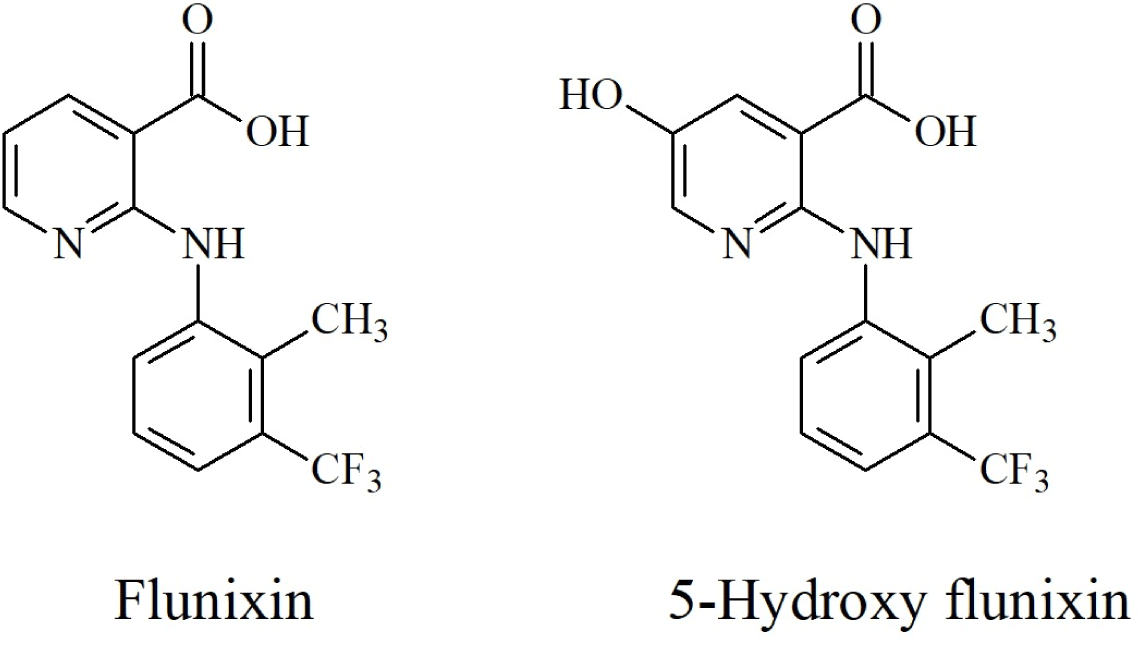
However, Method 1 is an easy and efficient method owing to its simpler sample preparation compared with Method 2. Method 1 is a multi-class, multi-residue analysis method used in Korea to determine the residues of 157 veterinary drugs in livestock products. In addition, Method 1 does not interrupt the existing multi-class, multi-residue analysis method. The structures of the analytes and their selectivities are shown in Figs. 1 and 2.
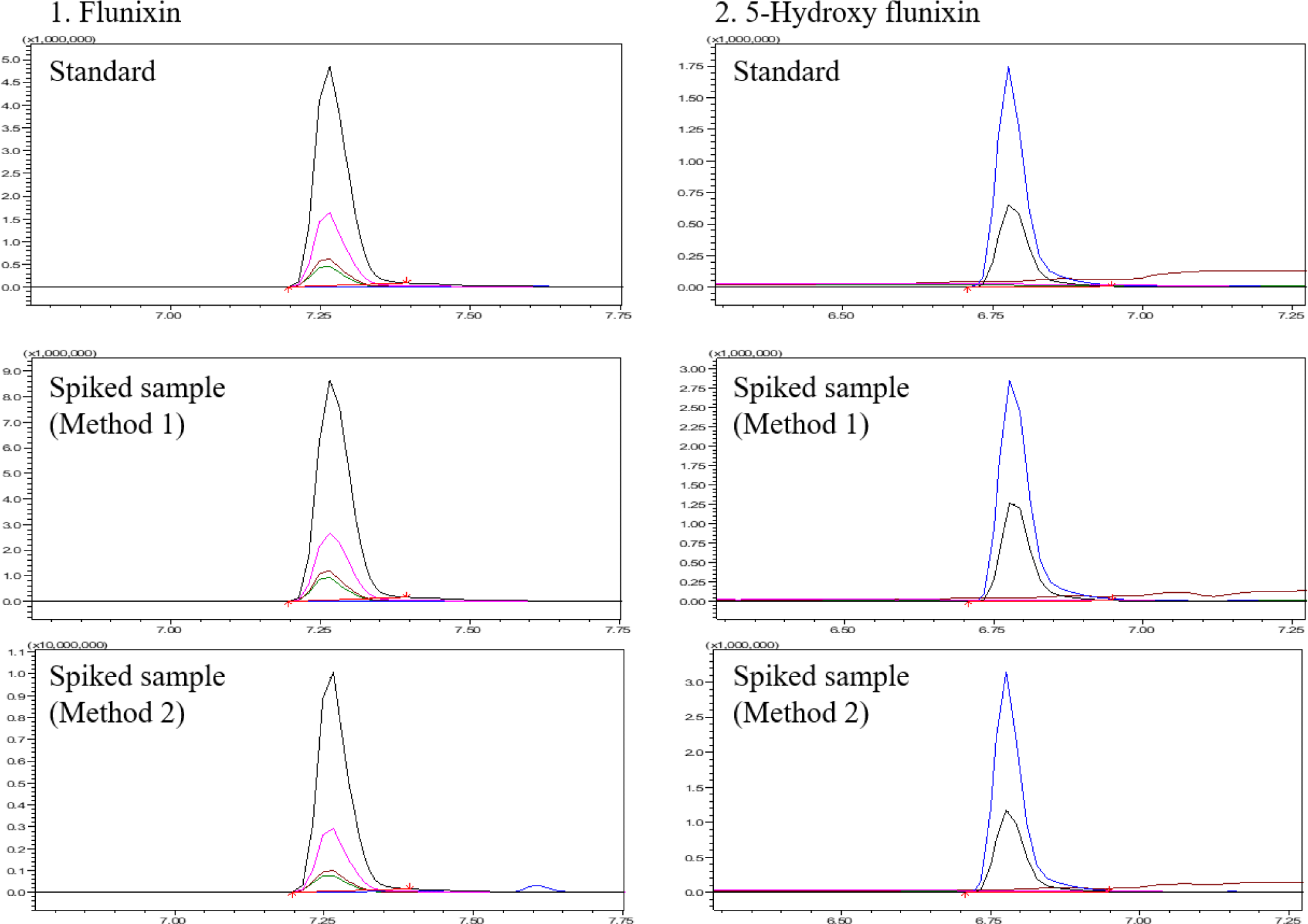
In Korea, the MRL of milk is 20 μg/kg for 5-hydroxy flunixin. In Method 1, the LODs and LOQs were 4 and 15 μg/kg for flunixin and 5 and 15 μg/kg for 5-hydroxy flunixin, respectively. In Method 2, the LODs and LOQs were 2 and 5 μg/kg for flunixin and 3 and 9 μg/kg for 5-hydroxy flunixin, respectively. Both methods can be used to quantify concentrations lower than the MRL. The matrix effects of flunixin and 5-hydroxy flunixin were –15% and –24.2% in Method 1 and 78.5% and 65.9% in Method 2, respectively (Table 4). Method 1 had no matrix effect on flunixin and a medium matrix effect on 5-hydroxy flunixin, whereas Method 2 had a strong matrix effect on both compounds. Considering the importance of quantifying 5-hydroxy flunixin in milk, both methods satisfied the Codex validation criteria. However, Method 1 has a simpler sample preparation and less matrix effect, and can be applied to multi-class, multi-residue analysis. Therefore, the present study proceeded with additional validation using other livestock and fishery products.
The inter-lab validation of Method 1 was assessed (Table 2). For flunixin and 5-hydroxy flunixin, the recoveries were 74.5%–94.0% and 71.2%–94.0%, and the inter-lab CVs were 8.9%–22% and 14%–20%, respectively. Method 1 was validated using intra- and inter-lab validation results that met the Codex guidelines (CAC/GL 71-2009). According to the guidelines, the recovery should be within the range of 60%–120% at 1–10 μg/kg and 70%–120% at 10–100 μg/kg. And the intra-lab and inter-lab CV should be less than 30% and 45% at 1–10 μg/kg and 20% and 32% at 10–100 μg/kg, respectively. In previous studies for milk or bovine muscle, ultrasound-assisted extraction showed 0.5 μg/kg of CCα for both flunixin and 5-hydroxy flunixin (Daeseleire et al., 2003) and 22.85 and 27.28 μg/kg of CCα and CCβ for flunixin (Lugoboni et al., 2014), respectively. Extraction with acetonitrile and wash with n-hexane showed 45.04 and 50.08 μg/kg of CCα and CCβ for 5-hydroxy flunixin, respectively (Malone et al., 2009). The present method used sorbents and washed the extract with n-hexane, as described in Method 1. These processes are necessary to improve purification, and filtration is required to eliminate particulates from the final sample (Malone et al., 2009). Acidic organic solvents, such as hydrochloric acid, were introduced for extraction, and the LODs were 2.98 μg/kg of flunixin in bovine muscle and 0.78 μg/L of 5-hydroxy flunixin in milk (Chen et al., 2019). For the bovine muscle sample, potassium acetate buffer (adjusted to pH 4.5 with acetic acid) and enzymatic hydrolysis using β-glucuronidase were also used with 6 mg/kg of LOD (Asea et al., 2001). Method 1 of the present study enabled the quantification of flunixin residues down to the MRL using the usual QuEChERS method, without the use of acids or buffers.
Method 1 was validated using other animal-based food products (Table 5). Five livestock and fishery products (beef, chicken, eggs, flatfish, and shrimp) were evaluated. All matrices showed good linearity (>0.99), and the recoveries and CVs were 82.4%–110% and 1.2%–8.9% for flunixin and 83.3%–106% and 2.2%–20% for 5-hydroxy flunixin, respectively. The matrix effects of Method 1 are listed in Table 4. For all matrices, except milk, Method 1 was estimated to have a strong matrix effect. Flunixin exhibited the strongest matrix effect in eggs, followed by shrimp, flatfish, beef, and chicken. 5-Hydroxy flunixin showed the strongest matrix effect in flatfish, followed by beef, chicken, eggs, and shrimp. When using LC-MS/MS, the matrix effect depends on the sample properties, ionization interface, mobile phase additives, stationary phase, and other characteristics. The ionization intensity can be reduced by nonvolatile substances via increasing the viscosity and surface tension and decreasing the solvent evaporation rate (Silvestro et al., 2013). Therefore, the strong matrix effect is interpreted as a phenomenon caused by ion suppression. Simple purification with n-hexane and C18 is not sufficient to reduce matrix effects, however, this can be addressed using a matrix-matched calibration.
Recently, the strict monitoring of veterinary drug residues has been emphasized with the introduction of the PLS system in Korea. Consistent with this situation, analysis methods for all animal-based foods are required. A previous FDA study optimized a multi-class, multi-residue LC-MS/MS method to determine 25 veterinary drug residues (Clark et al., 2011). In the abovementioned method, acetonitrile was added to extract target compounds and precipitate protein, and SPE using OASIS® HLB was also employed; however, this method is only for milk and requires additional confirmation to apply other animal-based food. Food analysis using SPE not only has the disadvantage of clogging the adsorbent but also requires conditioning and cleaning steps for activation.
The detection limits of Method 1 were assessed for six animal-based food products (milk, beef, chicken, egg, flatfish, and shrimp) and are described in Tables 2 and 5. The LODs and LOQs were 2–8 and 5–27 μg/kg for flunixin and 2–10 and 6–33 μg/kg for 5-hydroxy flunixin, respectively. However, Method 1 showed poor recovery of 5-hydroxy flunixin from pork muscle and eels. Recoveries and CVs of flunixin were 96.0%–98.9% and 2.9%–4.1% at 25, 50, and 100 μg/kg in pork and 91.0%–99.0% and 1.5%–9.1% at 5, 10, and 20 μg/kg in eel, respectively. In contrast, the recovery of the same concentration of 5-hydroxy flunixin from pork and eel was <40%. Therefore, Method 1 could be used for the quantification of flunixin or the identification of 5-hydroxy flunixin in pork and eels in a limited manner.
Most studies of flunixin residues have been conducted on beef or milk (Daeseleire et al., 2003; Douglas et al., 2012; Gallo et al., 2010; Kissell et al., 2013; Malone et al., 2009) and only a few have investigated swine and chicken (Chou et al., 2022; Liu et al., 2015). Flunixin is approved for swine at a dosage of 2.2 mg/kg with a withdrawal time of 12 d (Sidhu et al., 2017). In a previous flunixin study, recovery and repeatability were 76.7%–86.8% and 4.1%–12.5% in swine tissues and 73.8%–84.0% and 4.1%–9.9% in chicken tissues, respectively, and the LOQ ranged from 0.05 to 0.50 μg/kg in both matrices (Liu et al., 2015). However, the previous study was performed only for flunixin and used hydrolysis with hydrochloric acid and heating. Although the LOQs in the present study were higher than those of previous studies, the method is beneficial in terms of its simple process and application to other matrices for two compounds (flunixin and 5-hydroxy flunixin) in livestock and fishery products.
Conclusion
This study assessed the linearity, accuracy, precision, detection limits, and matrix effect of two methods for the analysis of flunixin and 5-hydroxy flunixin in milk. Two methods involve QuEChERS-based extraction and purification followed by analysis by LC-MS/MS. Both methods can quantify the two compounds at a lower level than their MRL in milk. Method 1 is the existing multi-class, multi-residue method used in Korea, and Method 2 is a little more complicated than Method 1, including the use of EDTA, formic acid, and dichloromethane. Applicability of Method 1 to four livestock and two fishery products (milk, beef, chicken, egg, shrimp, and flatfish) has been confirmed. The results of this study should improve the efficiency of the monitoring system and emphasize the importance of improving multi-class, multi-residue analysis methods. In future studies, improvements through additional purification processes are needed to obtain low detection limits and MEs even in simultaneous analysis methods.