









Introduction
Listeria monocytogenes is a gram-positive, facultative intracellular foodborne pathogen (Vàzquez-Boland et al., 2001). The pathogen is the causative agent of listeriosis in animals and humans, which is associated with high mortality, especially in humans (Vàzquez-Boland et al., 2001). In humans, listeriosis can cause meningitis, septicemia, and abortion (Locatelli et al., 2013). There are 13 L. monocytogenes serotypes, 1/2a, 1/2b, 1/2c, 3a, 3b, 3c, 4a, 4b, 4ab, 4c, 4d, 4e, and 7, and most human cases of listeriosis are caused by infection with serotypes 1/2a, 1/2b, and 4b (Kathariou, 2002). In the USA, the incidence of listeriosis was estimated to be 0.3 cases per 100,000 in 2005 (CIDRP, 2011), which is close to the EU incidence rate of 0.3 cases per 100,000 (Denny and McLauchlin, 2008). In addition, the estimated mortality rate for listeriosis in the EU was reported to be 16.6%, which is similar to that reported in the USA (EFSA, 2011). Thus, many countries have investigated the prevalence of L. monocytogenes and the serotypes present in various environments, including livestock farms, slaughterhouses, and meat-processing plants to elucidate the potential pathways of L. monocytogenes transmission.
L. monocytogenes is widely distributed in various environments, such as water and soil, and the pathogen can be transferred to animals. Animals infected with L. monocytogenes are generally asymptomatic carriers and can shed the pathogen in their feces, which may contaminate fresh and processed meat products (Boscher et al., 2012). Watkins and Sleath (1981) showed that the levels of L. monocytogenes in sewage sludge on contaminated farm land remained unchanged for at least eight weeks. Silage has also been identified as a risk factor for farm animal listeriosis through feed consumption, which can result in dissemination of the pathogen in farm environments through animal feces (Fenlon et al., 1996; Weidmann et al., 1996).
In France, Boscher et al. (2012) investigated the prevalence of L. monocytogenes and its serotypes in sows in 2008, and they showed that 46% of sow farms and 11% of the collected samples were positive for the pathogen. The serotype distribution was as follows: 1/2a (41%), 1/2b (36%), 4b (21%), and 1/2c (2%). In the feces of black beef cattle in Japan, the L. monocytogenes isolation rates were 11.4% of 651 samples (northern region), 2.8% of 572 samples (central region), and 2.9% of 515 samples (southern region). Moreover, 1/2b was the most prevalent serotype (40.5%) followed by 1/2a (36.9%), 4b (21.6%), and 4ab (1.0%) (Megumi et al., 2014). However, in Korea, the prevalence of L. monocytogenes on livestock farms has not been studied; thus, very limited information is available on the occurrence of L. monocytogenes.
Therefore, this study aimed to estimate the current prevalence of L. monocytogenes on livestock farms in Korea through molecular identification and serotyping of L. monocytogenes isolates and by analyzing the genetic correlations among the isolates by pulsed-field gel electrophoresis (PFGE).
Materials and Methods
The following samples were collected from each livestock farm: fecal (10), soil (10), silage (10) for pig and cattle farms, and sludge (1) only from pig farms. This same number of samples was collected at each visit from 25 farms (20 cattle farms and 5 pig farms). Each farm was visited 2-3 times in a 1-year period. Fecal samples were collected from animal pens with sterile specimen cups (SPL Life Science, Korea), and soil samples were collected from the soil around the animal pens with sterile specimen cups. Silage samples were collected in plastic sample bags, and sludge samples were collected in sterile specimen cups. All samples were placed on ice in a cooler and transferred to our laboratory for analysis.
To isolate L. monocytogenes from the samples, the method published by the Manufacturing and Ingredient Regulations for Livestock Products (MFDS, 2015) was used, with some modifications. Briefly, 25-g samples of feces, soil, or sludge were transferred from the specimen cups to sterile plastic sample bags containing 50 mL of Listeria enrichment broth (LEB; Becton Dickinson and Company, USA), and homogenized with a pummeler (BagMixer® 400; Interscience, France) for 60 s. To enrich L. monocytogenes in the samples, the homogenates were incubated at 30℃ for 24 h. Then, 1-mL aliquots of the enrichments were added to 9 mL of Fraser broth (Becton Dickinson and Company) and incubated at 37℃ for 24-48 h. A loopful of the Fraser broth was streaked on Palcam agar (Becton Dickinson and Company), and the plate was incubated at 30℃ for 48 h. Three to five L. monocytogenes-like colonies from each Palcam agar plate were inoculated into 10 mL of tryptic soy broth plus 0.6% yeast extract (TSBYE; Becton Dickinson and Company) and incubated at 30℃ for 24 h. A loopful of the cultures was then streaked on brain-heart infusion (BHI) agar (Becton Dickinson and Company) and incubated at 30℃ for 24 h. The colonies on BHI agar were used for identification by PCR.
The colonies on the BHI agar plates were analyzed by PCR (Rotor-GeneQ; Qiagen, Germany) for the presence of prs, a Listeria spp.-specific gene (Doumith et al., 2004), and hlyA, a L. monocytogenes-specific gene (Burall et al., 2011). The PCR amplification was carried out at 98℃ for 30 s, followed by 35 cycles of denaturation at 98℃ for 5 s, annealing at 60℃ for 5 s, and extension at 72℃ for 10 s, and a final 1-min extension at 72℃.The hlyA-positive samples were further analyzed by 16S rRNA gene sequencing to confirm their identity as L. monocytogenes. The genomic template DNA (20 ng) was amplified by EF-Taq (Solgent, Korea) in a 30 μL reaction mixture. An initial activation step was performed at 95℃ for 2 min, followed by 35 cycles of denaturation at 95℃ for 1 min, annealing at 55℃ for 1 min, and extension at 72℃ for 1 min, with a final extension step at 72℃ for 10 min. A multiscreen filter plate (Millipore Co., USA) was used to purify the amplified products and the primers 27F (5'-AGA GTT TGA TCM TGG CTC AG-3') and 1492R (5'-TAC GGY TAC CTT GTT ACG ACT T-3') (Lane et al., 1991; Weisburg et al., 1991). DNA samples containing the extension products were mixed with Hi-Di formamide (Applied Biosystems, USA). Then, the mixture was incubated at 95℃ for 5 min, on ice for 5 min, and then analyzed with an ABI Prism 3730XL DNA analyzer (Applied Biosystems).
Isolated L. monocytogenes strains were serotyped according to the method described by Doumith et al. (2004). Five primer sets (ORF2819, ORF2120, lmo0737, lmo1118, and prs) were used in a multiplex PCR. A single colony was suspended in 100 μL of the mixture of sterile distilled water and 50 μL of a 0.25% sodium dodecyl sulfate-0.05 N NaOH solution, and heated at 99℃ for 15 min. Then, 2 μL of the suspension was used as the DNA template. The concentration of each primer was 0.2 μM. Then, the template and primers were mixed in a reaction with a final volume of 50 μL using the Qiagen multiplex PCR kit (Germany). The multiplex PCR was performed in a Rotor-Gene Q (Qiagen), with an initial denaturation step at 94℃ for 15 min, followed by 35 cycles of denaturation at 94℃ for 30 s, annealing at 57℃ for 90 s, and extension at 72℃ for 60 s, with a final 30-min extension at 60℃. After PCR, 20 μL of the reaction mixture was mixed with 4 μL of loading buffer (Dyne Bio, Korea) and separated on a 1.5% agarose gel in 1× TAE buffer (Biosesang, Korea) for 20 min. Then, the serotypes of the L. monocytogenes isolates were determined by analyzing the banding patterns of the products from the multiplex PCR. Serotypes were also determined by an agglutination assay using Listeria antisera (Denka Seiken, Japan) according to the manufacturer’s instructions. When the multiplex PCR analysis and agglutination test results were in agreement, the serotypes of the L. monocytogenes isolates were confirmed.
To detect virulence genes, such as inlA, inlB, actA, plcB, and hlyA, typical L. monocytogenes colonies on Palcam agar were resuspended in 100 μL of sterile distilled water and 50 μL of 0.05 N NaOH-0.25% sodium dodecyl sulfate solution and incubated at 99℃ for 15 min. For virulence gene amplification by PCR, an aliquot of this mixture (2 μL) was mixed with the components of the Phire Hot Start II DNA Polymerase Kit (Thermo Fisher), Taq DNA polymerase mix (20 mM Tris-HCl, pH 7.4 at 25℃; 0.1 mM EDTA; 1 mM DTT; 100 mM KCl; 200 μg/mL BSA; and 50% glycerol), 1× reaction buffer (1.5 mM MgCl2), 200 μM deoxynucleoside triphosphates (dNTPs; Promega Co., USA), and 0.5 μM each virulence gene primer listed in Table 1. PCR was performed in a Rotor-Gene Q with the following program: an initial denaturation step at 98℃ for 30 s, followed by 35 cycles of 98℃ for 5 s, 60℃ for 5 s, and 72℃ for 10 s, with a final extension step at 72℃ for 1 min. After PCR, an aliquot of the reaction products (20 μL) was mixed with 4 μL of loading star, and then separated by electrophoresis in a 1.5% agarose gel.
| Primer | Size (bp) | Sequence (5′ to 3′) | Reference |
|---|---|---|---|
| inlA | 255 | For: CCTAGCAGGTCTAACCGCAC | Khelef et al., 2006 |
| Rev: TCGCTAATTTGGTTATGCCC | |||
| inlB | 146 | For: AAAGCACGATTTCATGGGAG | Yong et al., 2003 |
| Rev: ACATAGCCTTGTTTGGTCGG | |||
| hlyA | 174 | For: GCATCTGCATTCAATAAAGA | Riedo et al., 1994 |
| Rev: TGTCACTGCATCTCCGTGGT | |||
| actA | 385 | For: GACGAAAATCCCGAAGTGAA | Gianfranceschi et al., 1998 |
| Rev: CTAGCGAAGGTGCTGTTTCC | |||
| plcB | 261 | For: GGGAAATTTGACACAGCGTT | Winters et al., 1999 |
| Rev: ATTTTCGGGTAGTCCGCTTT |
To determine the genetic correlations among the L. monocytogenes isolates, they were analyzed by pulsed-field gel electrophoresis (PFGE) according to the standard protocol of Graves and Swaminathan (2001), with some modifications. In brief, L. monocytogenes isolates from culture were embedded in 1% SeaKem gold agarose (Cambrex, USA) and lysed with cell lysis buffer (50 mM Tris, 50 mM EDTA, 1% Sarcosine) and proteinase K at 54-55℃ for 2 h. The agarose-embedded samples were washed with TE buffer (10 mM Tris, 1 mM EDTA, pH 8.0) 7 times, and then digested with the restriction enzyme AscI (Enzynomics, Korea) at 37℃ for 2 h. The restricted DNA fragments were separated through a 1% SeaKem gold agarose gel in 0.5× tris borate EDTA (TBE) with a CHEF MAPPER XA system (Bio-Rad, USA) at 14℃, 6.0 V/cm, and 120° with a linear ramping factor of 2.2-54 s for 19 h. XbaI-digested Salmonella enterica-serotype Braenderup H9812 DNA was used as a size standard. After electrophoresis, the agarose gel was stained with SYBR gold (Molecular Probes, USA) and de-stained with dH2O for 1 h. Images of the banding patterns were captured using an LAS-3000 imaging system (Fujifilm, Japan), and the PFGE patterns were analyzed with Gelcompar II 6.6 software (Applied Maths, Belgium). Similarity clustering analyses of the isolates were performed by the unweighted pair group method with arithmetic mean (UPGMA) using the Dice correlation coefficient and 1% tolerance.
Antimicrobial susceptibility testing was performed according to the Clinical Laboratory Standards Institute (CLSI, 2014). A single colony of each L. monocytogenes isolate was inoculated into 10 mL of tryptic soy broth containing 0.6% yeast extract (TSBYE; Becton, Dickinson and Company). After incubation at 30℃ for 24 h, the culture was diluted with 5 mL of phosphate buffered saline (PBS, pH 7.4; KH2PO4, 0.2 g; Na2HPO4, 1.5 g; NaCl, 8.0 g; KCl, 0.2 g in 1 L of dH2O) to an OD620 of 0.2. A sterile swab was dampened with the cell suspension, and approximately 1-2×108 CFU/mL of bacterial inoculum was spread on the surface of a Muller-Hinton agar plate (Becton Dickinson and Company). The plates were then incubated at room temperature for 10-15 min to allow the plate to absorb the liquid. Then, antibiotic discs (Oxoid, UK) were placed on the surface of the agar with a multidisc dispenser (Oxoid). After incubation at 30℃ for 24 h, the diameters of the clear zones were measured. Antibiotic sensitivity (resistant, intermediate, and susceptible) was judged according to the standards of the Clinical Laboratory Standard Institute (CLSI, 2014). The following 12 antimicrobial agents (amounts) were used: gentamycin (10 μg), penicillin G (10 units), tetracycline (30 μg), spectinomycin (100 μg), kanamycin (30 μg), erythromycin (15 μg), tigecycline (15 μg), ampicillin (10 μg), streptomycin (10 μg), vancomycin (30 μg), chloramphenicol (30 μg), and rifampicin (5 μg) (Oxoid, UK).
Results
A total of 2,018 samples were collected, including 677 feces samples, 680 soil samples, 647 silage samples, and 14 sludge samples. All feces, silage and sludge samples were negative for L. monocytogenes. Only 3 of the samples were positive for L. monocytogenes, and all 3 were soil samples obtained from a single cattle farm in the south east region. Thus, 3 of the 2,018 (0.15%) samples from livestock farms in Korea were contaminated with L. monocytogenes, which is a low prevalence rate. In addition, the farm from where the contaminated samples were obtained was revisited, however, no L. monocytogenes was isolated.
The 3 L. monocytogenes isolates were examined by multiplex PCR and agglutination assays to determine their serotypes. Multiplex PCR analysis can be used to classify L. monocytogenes strains into 1 of 5 serotype groups, group 1 (1/2a and 3a), group 2 (1/2c and 3c), group 3 (1/2b, 3b, and 7), group 4 (4b, 4d, and 4e), and group 5 (other serotypes and Listeria spp.), by the presence/absence of lineage-specific and serogroup-specific sequences (Doumith et al., 2004). The multiplex PCR results showed that isolate FI-2 was group 1 (1/2a and 3a), whereas the other isolates, FI-1 and FI-3, were group 5 (other serotypes such as 4a, 4d, 4ab; Fig. 1). Since multiplex PCR only determines serotype group, an agglutination assay was also conducted, and the results are shown in Table 3. Based on the results of the multiplex PCR and agglutination assays, the serotypes of the L. monocytogenes isolates were determined. L. monocytogenes FI-1 and FI-3 were determined to be serotype 4ab, and L. monocytogenes FI-2 was determined to be 3a, which are 2 serotypes that are not commonly isolated from food products and humans (Huang et al., 2015; Zhang et al., 2007).
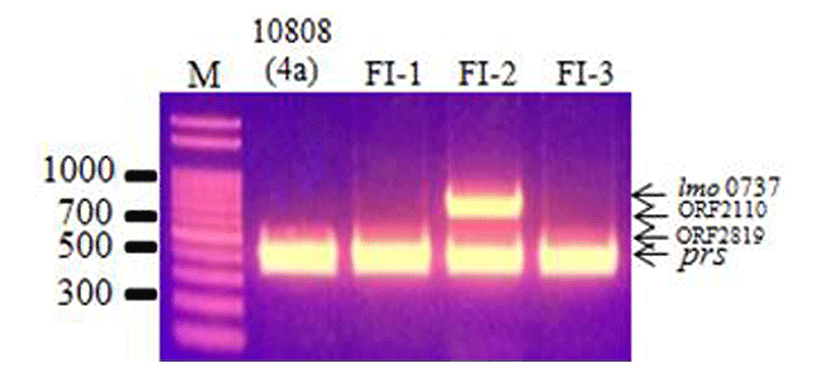
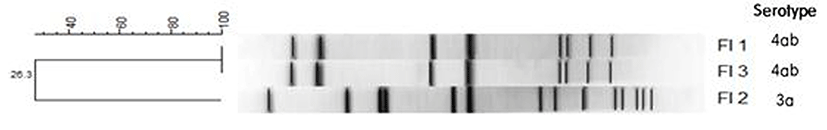
To determine the pathogenicity of the L. monocytogenes isolates, the presence of 5 virulence genes, actA, inlA, inlB, hlyA, and plcB, was detected by PCR. The FI-2 isolate showed all 5 virulence genes, indicating that it may be pathogenic to humans. The FI-1 and FI-3 isolates showed only actA, hlyA, and inlA, indicating that these isolates may have low pathogenicity (Wiedmann et al., 1996). The 3 L. monocytogenes farm isolates were genotyped using PFGE (Fig. 2). DNA macrorestriction with AscI suggests that the FI-1 and FI-3 isolates were 100% similar, whereas the FI-2 isolate (3a) shared only 26.3% similarity with the FI-1 and FI-3 isolates (4ab), showing low genetic correlation. These results show that although these strains of L. monocytogenes were isolated from the same geographical location, they had different serotypes and low genetic correlation.
Most L. monocytogenes isolates showed resistance to most tested antibiotics except tetracycline, erythromycin, and chloramphenicol. Of these 3 antibiotics, only isolate FI-3 was resistant to tetracycline and chloramphenicol. All 3 isolates had intermediate sensitivity to erythromycin (Table 3).
Values shown are the diameters (mean ± SD; mm) of clear zones.
A,BMeans within the same row with different superscript letters are significantly different (p<0.05).
R: resistant, I: intermediate, S: susceptible (CLSI, 2014)
Discussion
Although L. monocytogenes foodborne outbreaks have not been reported in Korea, many cases of L. monocytogenes infection have been reported. Thus, the potential sources of these L. monocytogenes infections need to be examined. Animal feces and soil are potential sources of L. monocytogenes (Oliver et al., 2005). Fox et al. (2009) reported that 14% and 25% of samples from silage and hay used for feeding and cattle bedding, respectively, were positive for L. monocytogenes. Silage contamination may cause infection in animals. Other studies (Ho et al., 2007; Schoder et al., 2011) showed variation in the prevalence of L. monocytogenes in feces. Thus, the pathogen may infect humans through consumption of contaminated meat. Although some studies isolated L. monocytogenes from slaughterhouses for hens, pigs, and cattle in Korea, the detection rate of L. monocytogenes on livestock farms was considerably lower than reported in other countries (Baek et al., 2000). Kang et al. (1998) and Yi et al. (2000) detected no L. monocytogenes contamination in fecal and water samples from livestock farms in Korea. Therefore, in this study, we examined potential contamination sources, such as feces, soil, silage, and sludge, on livestock farms, and we collected a total of 2,018 samples in 2-3 visits to each farm. Only 3 soil samples from a cattle farm in the south west region were contaminated with L. monocytogenes. Soil is considered a natural source of L. monocytogenes (Locatelli et al., 2013), and the pathogen may infect humans through food processing. The results of our study indicate that L. monocytogenes is present on livestock farms in Korea, which is in disagreement with previous data. However, the prevalence was very low compared to that in other countries such as France (11-46%) and Japan (2.8-11.4%) (Boscher et al., 2012; Megumi et al., 2014).
The serotypes of the L. monocytogenes isolates in this study were 3a and 4ab, which are rarely reported in food and human listeriosis (Doumith et al., 2004). Of the isolates, FI-2 is serotype 3a, which belongs to lineage II (Nadon et al., 2001), suggesting that, although serotype 3a is not a common cause of human listeriosis, it could cause human listeriosis. The finding of an isolate of this serotype is in agreement with the high prevalence of 3a serotypes on dairy farms (Jamali et al., 2013). The serotype 4ab L. monocytogenes isolates FI-1 and FI-3 do not belong to any lineage.
A recent study by Rahimi et al. (2010) suggested that Listeria species were resistant to tetracycline and penicillin G. Walsh et al. (2001) showed that 10.9% of L. monocytogenes isolates were resistant to 1 or more antibiotic agents, and were most frequently resistant to tetracycline (6.7%) and penicillin (3.7%). However, all the isolates in our study were multidrug resistant, and showed resistance to all tested antibiotics, including tetracycline. The high prevalence of resistance to tetracycline in these L. monocytogenes isolates may be caused by treating animal infections with tetracycline (Aureli et al., 2002; Vela et al., 2001). The high prevalence of L. monocytogenes multidrug resistance observed in our study, compared to that observed in other countries, is cause for concern; thus, control measures should be established to decrease antibiotic resistance. Although the 3 L. monocytogenes isolates were obtained from the same cattle farm, they were of 2 different serotypes, and the genetic correlation between these serotypes was very low. This suggests that L. monocytogenes strains of various serotypes that are genetically unrelated can be found in the same geographical region, which should be considered in epidemiological studies.
In conclusion, although the prevalence is low, L. monocytogenes can be isolated from livestock farms in Korea. The serotypes of the isolates were not those that commonly infect humans; however, the presence of virulence genes suggests that they could be infectious. In addition, the isolates showed resistance to multiple antibiotics, which is not common for L. monocytogenes. Therefore, control measures for L. monocytogenes on livestock farms are needed.