










Introduction
Lactobacilli are members of lactic acid bacteria (LAB) and are generally acid- and bile-tolerant (Herbel et al., 2013; Lebeer et al., 2008; Yuki et al., 1999). They are found in a variety of fermented foods and in the gastrointestinal tract of humans and animals (Mandal et al., 2016; Valeriano et al., 2017). Some Lactobacillus strains have also been reported to colonize the intestinal mucosal layer and epithelium of the host (Valeriano et al., 2017, Wang et al., 2009). Therefore, intake of Lactobacillus strains which express bioactive proteins in intestine in the form of fermented dairy foods is very attractive (Bermudez-Humaran et al., 2013).
There continues to be great interest in the development of genetic tools for the regulation of gene expression in lactobacilli. In particular, inducible promoters have proven to be powerful tools in many biotechnological areas. Indeed, several regulated expression systems for LAB have been reported in recent years, including a heat-shock inducible system (Maidin et al., 2014), sugar-inducible systems (Duong et al., 2011), bacteriocin-inducible systems (Axelsson et al., 2003; Sorvig et al., 2003), as well as other stresses (Benbouziane et al., 2013; Bohmer et al., 2013; Perez-Arellano and Perez-Martinez, 2003). One of the best characterised expression systems is the nisin-controlled gene expression (NICE) system in Lactococcus lactis, in which gene expression is activated by the addition of nisin in the culture medium (Horn et al., 2003), following which the L. lactis strain successfully secretes human interleukin-22 under regulation of the nisin-inducible promoter (Loera-Arias et al., 2014).
In the intestine, lactobacilli are exposed to bile, which facilitates fat absorption during digestion. Some lactobacilli can withstand the physiological concentrations of bile to survive or colonise the gut (Hamon et al., 2012; Taranto et al., 2006), and several groups have characterized gene expression in intestinal lactobacilli under gastrointestinal conditions (Alcantara and Zuniga, 2012; Chen et al., 2017; Pajarillo et al., 2015; Pfeiler et al., 2007). Previously, a gene expression system using a bile-responsive element located upstream of the bile-inducible transporter was developed in Bifidobacterium longum to induce protein expression in the intestinal tract (Ruiz et al., 2012). However, to the best of our knowledge, Lactobacillus expression system induced by bile, an intestinal signal, has not yet been developed.
Our previous proteomic study on the bile response of Lactobacillus johnsonii PF01 identified highly expressed proteins by bile (Lee et al., 2013). In this study, we selected promoter regions of the genes upregulated by bile and compared their bile-responsiveness using β-glucuronidase reporter gene. Herein, we report the development of a bile-responsive expression system for Lactobacillus plantarum.
Materials and Methods
The bacterial strains and plasmids used in this study are listed in Table 1. Escherichia coli strains were grown in Luria-Bertani broth (Difco, Petroit, MI, USA) at 37°C with shaking, while Lactobacillus strains were grown in De Man, Rogosa, and Sharpe (MRS) medium (Difco, USA) at 37°C without shaking. E. coli DH5α was used as a host strain for vector construction. When appropriate, ampicillin and erythromycin (Sigma-Aldrich, St. Louis, MO, USA) were used at concentrations of 100 μg/mL and 3 μg/mL, respectively, for selection of transformants of E. coli and Lactobacillus.
General procedures for DNA manipulation were performed as described previously (Sambrook et al., 1989). All enzymes were purchased from Takara Bio, Inc. (Japan). E. coli plasmid DNA was isolated using a QIAprep spin miniprep kit (Qiagen, Valencia, CA, USA) and Lactobacillus genomic DNA was isolated according to the method of Walker and Klaenhammer (1994).
E. coli DH5a transformation was done by heat shock method performed as described by Sambrook et al. (1989). Preparation of electrocompetent Lactobacillus cells and electrotransformation were done as described by Kim et al. (2005), with some modifications. Lactobacillus cells were grown in MRS broth supplemented with 1% glycine at 37°C until it reached early-log phase (OD600=0.2–0.3) and placed on ice for 10 min. Afterwards, they were washed twice in cold washing buffer (5 mM sodium phosphate, 1 mM MgCl2, pH 7.4), and resuspended in ice-cold electroporation buffer (1 M sucrose, 3 mM MgCl2, pH 7.4). On the other hand, 1 μg of plasmid DNA was added to 50 μL of ice-cold cell suspension (~109 CFU/mL) in a disposable cuvette (Gene Pulser® Cuvette, 0.2 cm electrode gap; Bio-Rad, Hercules, CA, USA) and held on ice for 5 min. This mixture was subjected to electroporation using GenePulser Xcell™ (BioRad, USA) using 200 Ω, 2.0 kV and 25 μF capacitance conditions. Finally, the cell suspension was spread on MRS agar plate supplemented with 3 μg/mL of erythromycin and then incubated at 37°C.
For bile induction experiments, recombinants were grown in MRS-erythromycin broth, harvested, and resuspended in MRS-erythromycin broth supplemented with bile (Bile bovine; Sigma-Aldrich, USA).
Among the genes upregulated by bile in L. johnsonii (Lee et al., 2013), four genes, which encode phosphoenolpyruvate-dependent sugar phosphotransferase, mannose-specific (EIIDMan), L-lactate dehydrogenase (LDH), HPr kinase (HPrK), and D-alanine-D-alanine ligase (DDL), were chosen for this study (Supplementary Table 1). The potential Pribnow box and –35 region of the promoters were predicted from the complete genome sequences of L. johnsonii PF01 (GenBank accession no: PRJNA67469), using BPROM and Neural Network Promoter Prediction tools at 80%–90% confidence, with a consensus length of 0.5-kb for each promoter sequence (Supplementary Table 2). Primers specific to the sequences of each promoter were designed (Supplementary Table 3) and polymerase chain reaction (PCR) amplification was performed using Pfu DNA polymerase with a thermal cycler (Takara, Japan) using standard procedures.
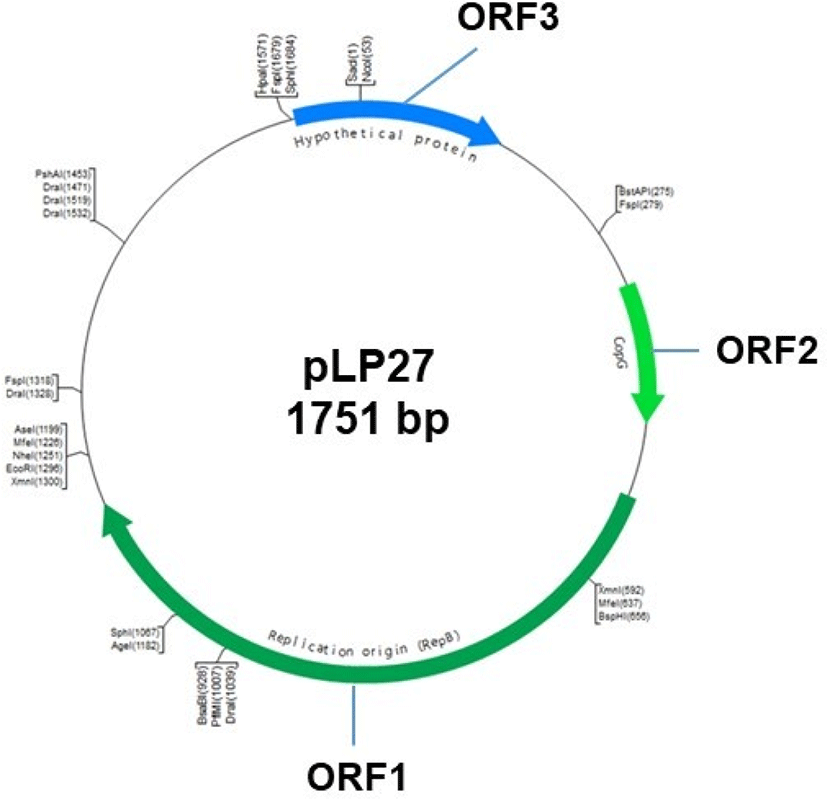
Plasmid pLP27 (Fig. 1) was isolated from L. plantarum LP27 from fermented cabbage for use as the backbone plasmid. Plasmid DNA was extracted using the standard alkaline lysis method (Sambrook et al., 1989) with modifications and purified using a gel extraction kit (Qiagen, USA). Purified pLP27 was digested with the restriction enzyme SacI and cloned into pUC19 for sequencing. Nucleotide sequence similarity searches and open reading frame (ORF) analysis were performed using the BLAST program at the NCBI site (http://www.ncbi.nlm.nih.gov/BLAST/).
The primers to clone the promoter regions for this study are listed in Supplementary Table 3. Erythromycin resistance gene as a selection marker was amplified by PCR from the genome of L. johnsonii PF01 with the primers Emr-F and Emr-R and cloned into the KpnI site of plasmid pULP1, yielding the pULP2, as shown in Fig. 2. Next, promoter sequences and the reporter gene β-glucuronidase (gusA) were amplified from the genome of L. johnsonii PF01 and pNZ8008, respectively, and fused via a second round of PCR. The resulting product was inserted into the SalI and PstI sites of pULP2, generating pULP3-xxx (xxx: PEIIDM, PLDH, PHPrK, or PDDL), which contain different kinds of promoters, respectively. Finally, the constructed plasmids were introduced into L. plantarum SK156 to measure promoter activity.

The transformants were selected and incubated in MRS broth containing erythromycin (3 μg/mL). After growth to mid-log phase (A600=0.4–0.5), cells were harvested by centrifugation at 4°C and washed with ice-cold MRS broth. Then they were resuspended in an equal volume of MRS broth supplemented with bile (0%–0.1% (w/v)) and erythromycin (3 μg/mL), and incubated at 37°C for 1 h. Cell-free extracts (CFEs) were harvested following cell disruption by sonication. Protein concentrations were determined using the Bio-Rad protein assay kit (Bradford, 1976). Promoter activity was assessed according to the GusA activity of CFEs, which was measured by the hydrolysis of 4-nitrophenyl β-D-glucuronide (PNPG; Sigma-Aldrich) as described previously (Platteeuw et al., 1994). For assay, CFEs were warmed to 37°C and 200 μL of CFE was added to 800 μL of GUS buffer (100 mM sodium phosphate, 2.5 mM EDTA, and 1.0 mM PNPG, pH 6.0). After incubation at 37°C for 5 min, 1 mL of 0.2 M Na2CO3 was added to stop the reaction. The units of Gus activity are described as picomoles of 4-nitrophenyl liberated per minute per milligram of protein. Three biological replicates were performed for all experiments.
The segregational stability of the construct pULP3-xxx was evaluated in plasmid-free L. plantarum SK156. Transformed L. plantarum SK156 was grown in MRS medium without erythromycin for approximately 120 generations. Every 20 generations, an aliquot of the culture was collected, spread on erythromycin-free MRS agar medium, and 100 colonies were picked and replicated on MRS agar medium with and without erythromycin. Colonies grown on each medium were counted and calculated as follows: (Ne/N)×100, where Ne and N are the number of colonies grown on MRS medium with and without erythromycin, respectively. Three biological replicates were performed for all experiments and data are represented as mean±SEM.
Results and Discussion
Plasmid pLP27 is a native plasmid of L. plantarum LP27 originally isolated from fermented cabbage. This plasmid was linearized with the restriction enzyme SacI and cloned into vector pUC19, generating pULP1. DNA sequencing of pLP27 revealed a 1,751 bp product with a GC content of 41.2% and three putative ORFs (Fig. 1). The products of the three ORFs and the most homologous protein of each identified in the GenBank database are listed in Table 2. The translated protein sequence of ORF1 showed that it shares 93% identity with the CopG family transcriptional regulator of L. heveticus DSM 20775. CopG is the prototype of a series of repressor proteins that are encoded by plasmids exhibiting a similar genetic structure at their leading strand initiation and control regions (del Solar et al., 1995). CopG also regulates plasmid copy number by binding the replication protein promoter for CopG (Gomis-Ruth et al., 1998; Hernandez-Arriaga et al., 2009). Therefore, ORF1 may be a transcriptional regulator for the expression of pLP27. ORF2 encoded a protein of 219 amino acids with 98% identity with the replication protein RepB of plasmid pLH2 of L. helveticus ATCC15009, which belongs to the rolling-circle replication plasmid family (van Kranenburg et al., 2005; Zhai et al., 2009). This indicates that pLP27 may replicate by a rolling-circle mechanism, which should be elucidated in the future. The predicted protein of ORF3 had no conserved domains or homolog sequences in the database, indicating that it is a unique, hypothetical protein. Plasmid pLP27 combined with pUC19, namely pULP1, was used as a backbone for the construction of bile-responsive expression vectors.
Our previous proteomic analysis of L. johnsonii PF01 revealed some proteins were upregulated by bile (Lee et al., 2013). Among the genes upregulated above two-fold at protein level by bile, genes encoding EIIDMan, LDH, HPrK, and DDL were selected and their promoters were used to construct bile-responsive expression vectors (Supplementary Table 1). Fig. 2 shows the cloning steps for construction of the pLP27-derived bile-responsive expression vectors. First, the erythromycin resistance gene from L. johnsonii PF01 was cloned into the KpnI site of pULP1, generating pULP2. The promoter regions of four genes were amplified, respectively, from the genome of L. johnsonii PF01 and fused upstream of the gusA reporter gene. Then, these constructs were cloned into the SalI and PstI sites of pULP2, generating pULP3-PEIIDM, pULP3-PLDH, pULP3-PHPrK, and pULP3-PDDL, respectively (Fig. 2).
Once all of the constructs were obtained, they were introduced by electroporation into L. plantarum SK156 (Table 1), and GusA activity was measured in different bile concentrations (Kim et al., 2006). The promoterless vector, which contains the gusA gene only was used as a negative control. Transformants were grown to early stationary phase either in the presence or absence of bile. Promoter activity was analysed by measuring GusA activity using PNPG as a substrate. As shown in Fig. 3, the LDH promoter showed the highest β-glucuronidase activity in L. plantarum and its activity increased 1.8-fold by bile addition. EIIDM and HPRK promoters were also inducible by bile, although their activities were lower than LDH promoter. On the other hand, DDL promoter was not responsive to bile.
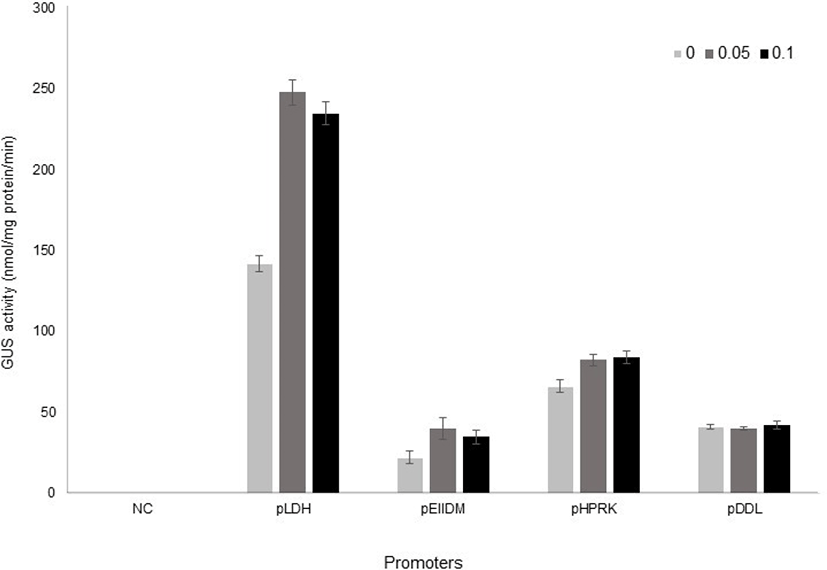
Interestingly, the expression of EIIDM was more inducible by bile than that of LDH in L. johnsonii (Supplementary Table 1). A report by Ruiz et al. (2012) showed that a bile-responsive expression system responded differently according to bifidobacterial species, suggesting that host difference might cause different expression level. In addition, we cannot exclude the possibility that the bile responsiveness of these promoters also varies according to bile composition, because arabinofuranosidase activity as a reporter in Bifidobacteria was strongly induced by cholate and conjugated cholate, but not by other bile salts (Ruiz et al., 2012). The activity of promoters used in this study according bile salts also need to be elucidated in the future.
Interestingly, GusA activities in all constructs was observed without bile, although their levels except for DDL promoter were lower than in the presence of bile. It indicates that the promoter’s strength can be regulated by other factors, too.
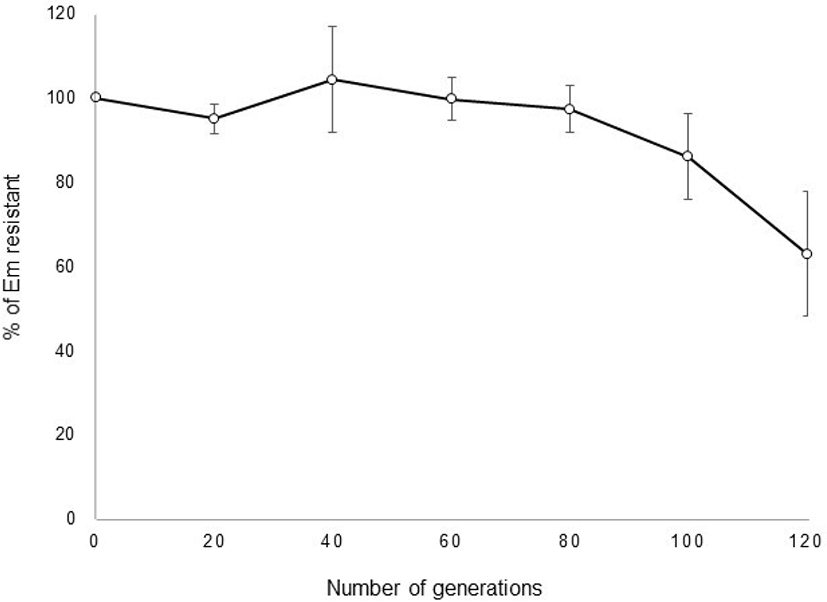
pULP3-PLDH showing the highest GusA activity was used to investigate segregational stability in the L. plantarum. The transformation efficiency was 1.2×104 CFU/μg DNA in L. plantarum SK156 (data not shown). It was also successfully introduced into L. fermentum. However, no transformants of pULP3-PLDH were obtained from L. acidophilus, L. johnsonii, and L. reuteri (data not shown). These results indicate that this vector has a limited host range. After selection of recombinant transformants, the stability of pULP3-PLDH in L. plantarum SK156 was monitored over 120 generations of growth in MRS broth. No loss of the plasmid was observed until 80 generations in the absence of erythromycin, and plasmid possession ratio decreased gradually to 63% after 120 generations (Fig. 4), indicating relatively high stability of the plasmid vector without selection pressure.
Conclusion
Although there continues to be great interest in the development of genetic tools for the regulation of gene expression in lactobacilli, to the best of our knowledge, a bile-responsive expression system in lactobacilli has not yet been developed. Herein, we report the development of a bile-responsive expression system for Lactobacillus plantarum. This system can be an effective tool for the expression of bioactive proteins in intestine after intake of the Lactobacillus spp. in the form of fermented dairy food.