










Introduction
Probiotics are live microorganisms that are well known to bring beneficial effects to the host and have been extensively studied as a therapeutic intervention by alleviating intestinal inflammatory responses and normalizing gut mucosal dysfunction (Borchers et al., 2009). Previous studies have demonstrated that not only whole live bacteria of probiotics but also cell wall components, bacterial lysates, enzymes, and amino acids from probiotics exert immunomodulatory effects (Izuddin et al., 2020; Kaji et al., 2010; Żółkiewicz et al., 2020). Genomic DNA of probiotics has also been recognized as a component that is capable of modulating immune responses. Indeed, genomic DNA extracted from Bifidobacterium and Lactobacillus has been shown to stimulate the production of interleukin (IL)-1β, IL-6, and IL-10 in human peripheral blood mononuclear cells (Lammers et al., 2003). Additionally, genomic DNA derived from Lactiplantibacillus plantarum is known to inhibit Escherichia coli lipopolysaccharide (LPS)-induced inflammatory responses in THP-1 cells (Kim et al., 2012).
Oral diseases impose a huge economic burden worldwide, with periodontitis representing one of the most common dental infections (Tonetti et al., 2017). Periodontitis is known as a chronic inflammatory disease present in tissues supporting the tooth, which destroys the alveolar bone and may result in tooth loss (Könönen et al., 2019). The pathogenesis of periodontitis is largely attributed to Porphyromonas gingivalis, a Gram-negative anaerobic bacteria that expresses several virulence factors (Mysak et al., 2014). Among these factors, P. gingivalis LPS is considered a key pathogenic component of periodontal diseases (Wang and Ohura, 2002). P. gingivalis LPS, which is recognized by Toll-like receptor (TLR)2 and TLR4, activates inflammatory signaling pathways and thus various pro-inflammatory cytokines and chemokines, including IL-1β, IL-6, and monocyte chemoattractant protein-1 (MCP-1; Darveau et al., 2004; Kato et al., 2014; Zhou et al., 2005).
Many previous studies have shown that probiotics, such as Lactobacillus spp., can reduce inflammatory responses and alleviate oral diseases (Gatej et al., 2018; Zhao et al., 2012). However, side effects have been reported for probiotics, including systemic infections and excessive immune stimulation (Doron and Snydman, 2015). Therefore, bioactive compounds from probiotics, known as postbiotics, have been introduced as novel alternatives (Moradi et al., 2020). In this study, we aimed to assess the postbiotic potential of genomic DNA from the probiotic strain Lactiplantibacillus plantarum against periodontitis by evaluating the anti-inflammatory effects of L. plantarum genomic DNA on P. gingivalis LPS-induced inflammatory responses.
Materials and Methods
L. plantarum ATCC 14917 was obtained from the American Type Cell Culture (ATCC; Manassas, VA, USA) and cultured as described previously (Kim et al., 2022). Genomic DNA of L. plantarum (LpDNA) was extracted using a Wizard Genomic DNA Purification Kit (Promega, Madison, WI, USA) following the manufacturer’s instructions with slight modifications. Briefly, bacteria were harvested by centrifuging and extensively washed with phosphate-buffered saline. The pellets were resuspended in 50 mM EDTA and lysozyme (10 mg/mL) followed by incubation at 37°C for 1 h. After centrifugation to obtain lysates, nuclei lysis solution (600 μL) was added to the lysates and incubated at 80°C for 5 min. Subsequently, RNase solution (3 μL) was added to the lysates and further incubated at 37°C for 40 min. Thereafter, the lysates were mixed with protein precipitation solution (200 μL), incubated on ice for 5 min, and centrifuged. After centrifugation, the supernatants were obtained and mixed with 600 μL of isopropanol and centrifuged as described above. The remaining lysates were washed with 70% ethanol, rehydrated by adding DNA rehydration solution, and incubated for 1 h at 65°C. The concentration and purity of LpDNA were measured using a NanoDrop spectrophotometer (ND-1000, NanoDrop Technologies, Hampton, NH, USA).
RAW 264.7, the murine macrophage cell line, was purchased from the ATCC (Manassas, VA, USA) and maintained in Dulbecco’s modified Eagle’s medium (DMEM; Welgene, Gyeongsan, Korea) supplemented with 10% heat-inactivated fetal bovine serum (Gibco, Burlington, ON, Canada), 100 U/mL penicillin, and 100 μg/mL streptomycin (HyClone, Logan, UT, USA) at 37°C in a humidified incubator under 5% CO2.
RAW 264.7 cells were plated in a 12-well culture plate and grown at 37°C for 24 h. The cells were pretreated with LpDNA (1 μg/mL) for 3, 9, or 15 h and subsequently stimulated with P. gingivalis LPS (PgLPS; 1 μg/mL) for a further 3 h. Total RNA was extracted and complementary DNA (cDNA) was synthesized with total RNA, random hexamers, and reverse transcriptase (Promega). cDNA was amplified by qPCR using a StepOnePlusTM Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) with SYBR Green Real-Time PCR Master Mix (Toyobo, Osaka, Japan). The amplification was conducted as follows: denaturation at 95°C for 10 s and amplification for 40 cycles at 95°C for 15 s and 60°C for 31 s. The sequences of specific PCR primers used in the study were as follows: IL-1β, forward, 5′-CTCACAAGCAGAGCACAAGC-3′ and reverse, 5′-TCTTGGCCGAGGACTAAGGA-3′; IL-6, forward, 5′-TCCTACCCCAATTTCCAATGCT-3′ and reverse, 5′-TCTGACCACAGTGAGGAATGTC-3′; MCP-1, forward, 5′-AGCCAACTCTCACTGAAGCC-3′ and reverse, 5′-TCTCC AGCCTACTCATTGGGA-3;′ TLR2, forward, 5’-CTGCGAAGTGGAAACCATCC-3′ and reverse, 5′-GCACTTCTGTTCC TCGCAGA-3′; TLR4, forward 5′-ACTCAGCAAAGTCCCTGATGAC-3′ and reverse, 5′-AGTTTGAGAGGTGGTGTAAGCC-3′; and β-actin, forward, 5′-TACAGCTTCACCACCACAGC-3′ and reverse, 5′-GGAAAAGAGCCTCAGGGCAT-3′. The relative mRNA expression of IL-1β, IL-6, MCP-1, TLR2, and TLR4 genes was normalized to that of β-actin using the 2−ΔΔCt method.
RAW 264.7 cells were plated in a 48-well culture plate and incubated at 37°C for 24 h. The cells were pretreated with LpDNA (1 μg/mL) for 15 h and stimulated with PgLPS (1 μg/mL) for a further 24 h. The secretion of IL-1β, IL-6, and MCP-1 in the spent culture supernatants was analyzed using commercial ELISA kits (R&D Systems, Minneapolis, MN, USA).
RAW 264.7 cells were incubated at 37°C for 24 h and pretreated with LpDNA (1 μg/mL) for 15 h and stimulated with PgLPS (1 μg/mL) for a further 30 min. Cell lysates were extracted using a lysis buffer containing enzyme inhibitors. Samples were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and electrotransferred onto polyvinylidene difluoride membranes (Millipore, Bedford, MA, USA). After blocking the membranes with 5% skimmed milk in Tris-buffered saline containing 0.1% Tween 20, the membranes were incubated with primary antibodies specific for extracellular signal-regulated kinase (ERK), phosphorylated ERK, p38 kinase, phosphorylated p38 kinase, c-Jun-N-terminal kinase (JNK), phosphorylated JNK, IκBα (Cell Signaling Technology, Danvers, MA, USA), or β-actin (Santa Cruz Biotechnology, Santa Cruz, CA, USA) at 4°C overnight. The membranes were incubated with horseradish peroxidase-conjugated anti-rabbit IgG (Cell Signaling Technology) or anti-mouse IgG (Santa Cruz Biotechnology) for 2 h at room temperature. Immunoreactive bands were detected and analyzed with an enhanced chemiluminescence reagent (Dyne Bio, Seongnam, Korea) and C-DiGit blot scanner (LI-COR Biosciences, Lincoln, NE, USA).
Results
To investigate whether LpDNA inhibits PgLPS-induced IL-1β, IL-6, and MCP-1 mRNA expression, RAW 264.7 cells were pretreated with LpDNA for 3, 9, or 15 h and subsequently stimulated with PgLPS for 3 h. As expected, PgLPS alone significantly induced IL-1β, IL-6, and MCP-1 mRNA expression. Although pretreatment with LpDNA for 3 h moderately inhibited PgLPS-induced IL-1β mRNA expression, LpDNA did not significantly inhibit PgLPS-induced IL-6 and MCP1 mRNA expression (Fig. 1A). Similarly, pretreatment with LpLTA for 9 h inhibited IL-1β mRNA expression induced by PgLPS, but not IL-6 and MCP-1 mRNA expression (Fig. 1B). However, pretreatment with LpDNA remarkably inhibited PgLPS-induced mRNA expression of those inflammatory mediators (Fig. 1C). Moreover, when RAW 264.7 cells were cotreated with LpDNA and PgLPS, LpDNA failed to inhibit PgLPS-induced IL-1β, IL-6, and MCP-1 mRNA expression (data not shown), suggesting that pretreatment with LpDNA for 15 h effectively inhibited PgLPS-induced inflammatory responses. Further experiments were conducted to investigate the anti-inflammatory effect of LpDNA after 15 h of pretreatment.
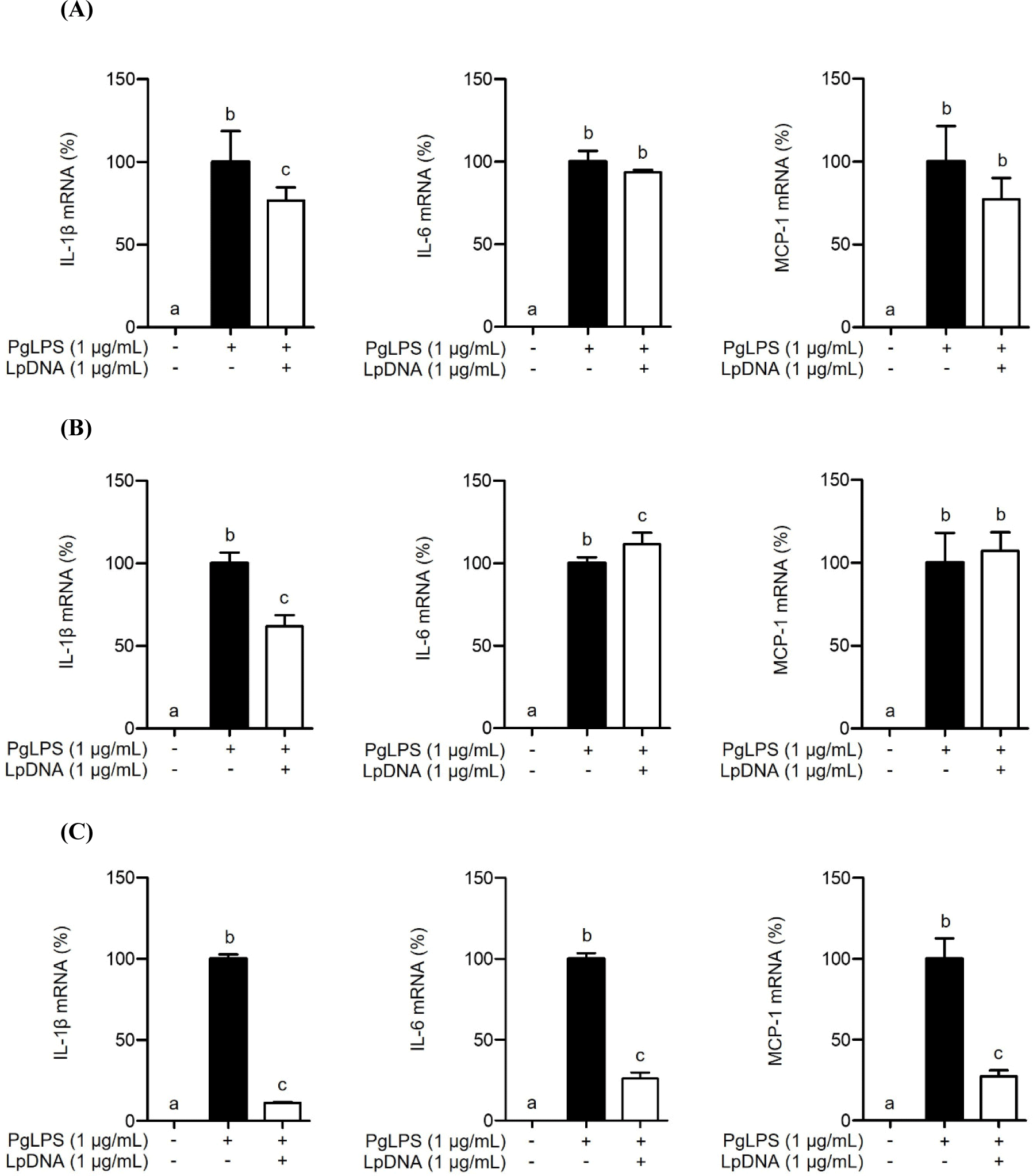
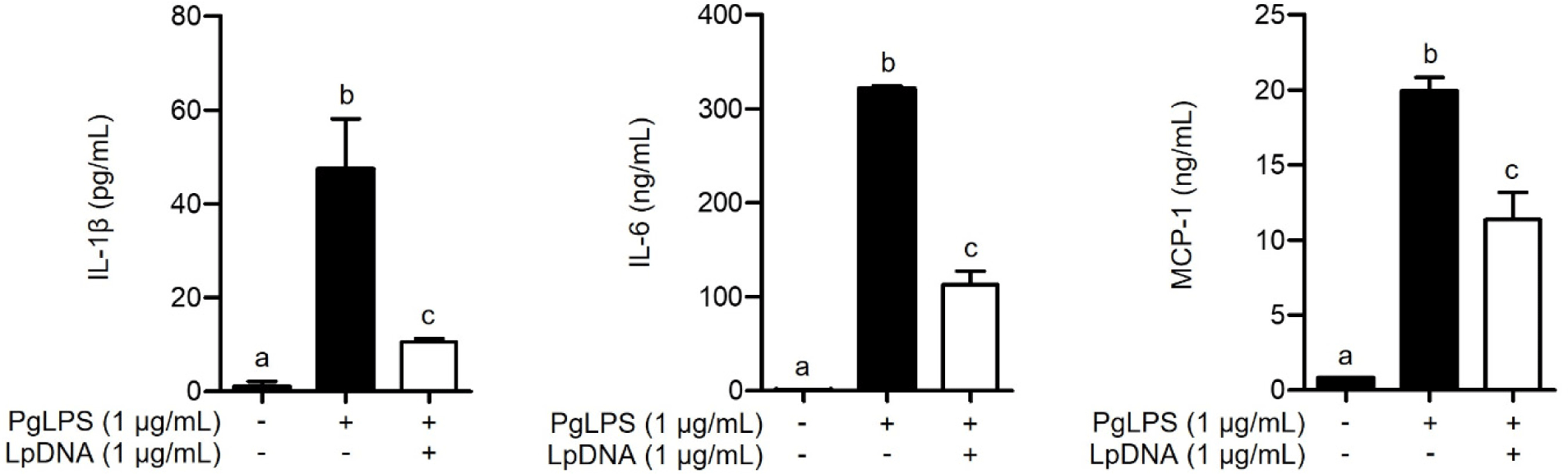
To investigate whether LpDNA inhibits the PgLPS-induced IL-1β, IL-6, and MCP-1 protein secretion, RAW 264.7 cells were pretreated with LpDNA for 15 h and subsequently stimulated with PgLPS for 24 h. As shown in Fig. 2, PgLPS significantly increased the protein secretion of IL-1β, IL-6, and MCP-1 in the absence of LpDNA pretreatment. However, IL-1β, IL-6, and MCP-1 protein secretion was dramatically decreased by pretreatment with LpDNA. These results suggest that LpDNA effectively inhibits PgLPS-induced inflammatory responses.
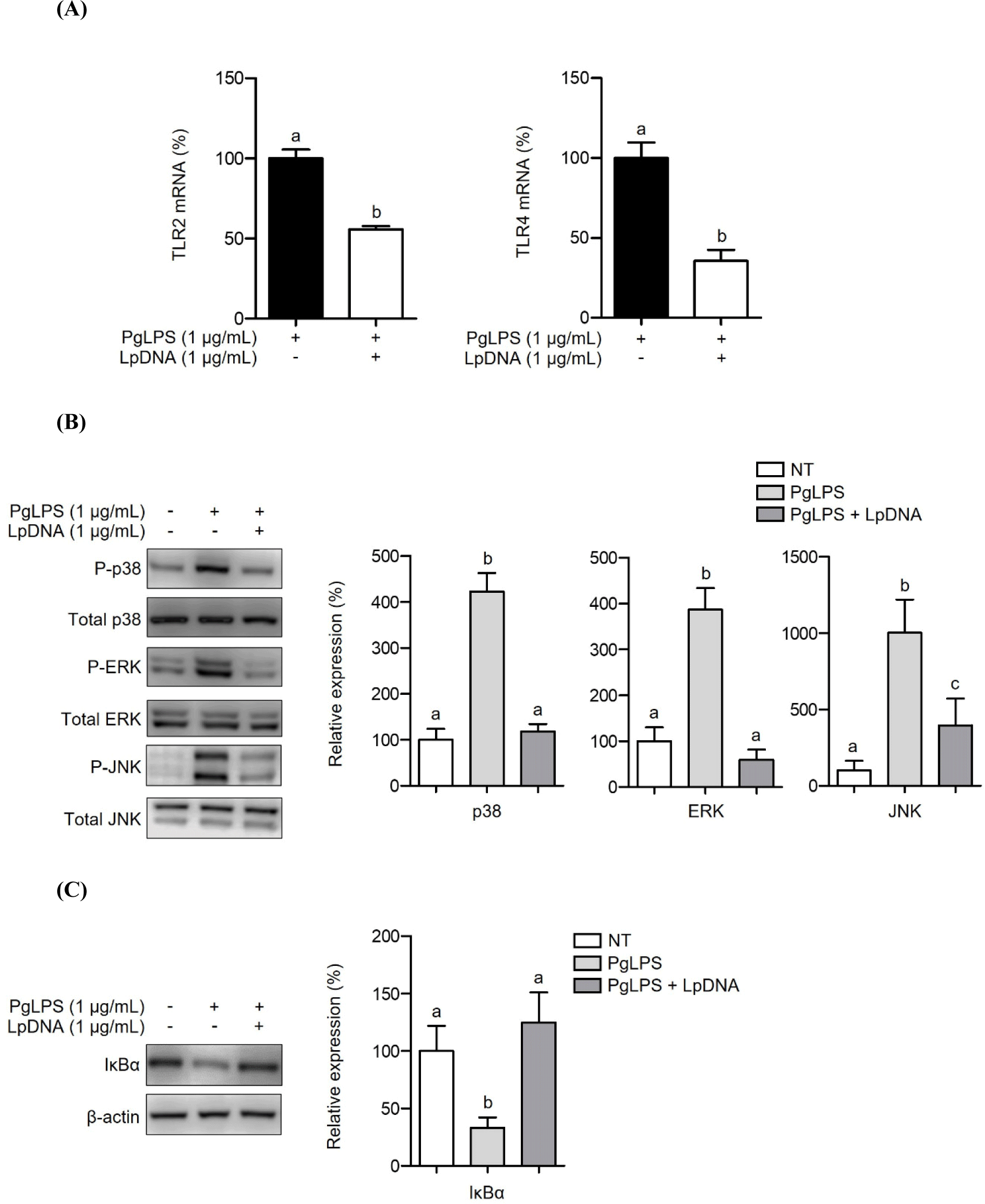
Both TLR2 and TLR4 are known to be simultaneously activated following recognition of PgLPS, resulting in the induction of inflammatory responses through downstream signaling pathways (Kocgozlu et al., 2009). Therefore, we examined whether LpDNA attenuates TLR2 and TLR4 expression. After pretreating RAW 264.7 cells with LpDNA and stimulating with PgLPS for 3 h, both TLR2 and TLR4 were significantly downregulated (Fig. 3A). TLRs recognize their ligands and trigger intracellular signal cascades to mount acute inflammatory responses (Wang and Ohura, 2002). The MAPK signaling pathways, including p38 kinase, ERK, and JNK, and the NF-κB signaling pathway are essential signaling pathways of inflammation that induce the secretion of proinflammatory cytokines (e.g., IL-1β) and subsequent inflammation (Kim et al., 2004). Hence, the activation of MAPKs and NF-κB was evaluated in RAW 264.7 cells pretreated with LpDNA in the presence of PgLPS. As shown in Fig. 3B, PgLPS alone increased the phosphorylation of p38 kinase, ERK, and JNK, while LpDNA pretreatment significantly reduced the phosphorylation of p38 kinases, ERK, and JNK. Moreover, an inhibitor of NF-κB, IκBα, which downregulates NF-κB activation (Wang et al., 2020), was degraded by PgLPS treatment alone, while LpDNA pretreatment restored IκBα in the presence of PgLPS (Fig. 3C). These results suggest that LpDNA-mediated TLR suppression downregulates the signaling cascades associated with PgLPS-induced inflammatory responses.
Discussion
Many studies have demonstrated that probiotics, in particular lactobacilli, exert beneficial effects, including antimicrobial, immune modulating, and blood cholesterol reducing activities (Duary et al., 2012; Kõll-Klais et al., 2005; Kwak et al., 2014). Lactobacilli also play an important role in attenuating periodontal diseases. However, the beneficial effects of structural components derived from L. plantarum, such as genomic DNA, have not been widely documented. Although a previous study has reported that genomic DNA of Pediococcus acidilactici suppressed P. gingivalis LPS-induced inflammatory responses (Choi et al., 2023), we investigated the inhibition of genomic DNA derived from L. plantarum on P. gingivalis LPS-induced inflammatory responses.
Pro-inflammatory cytokine production is known to be elevated in gingival crevicular fluid (GCF) where it facilitates bone resorption, resulting in loss of connective tissue attachment (Graves, 2008). The chemokine MCP-1 plays a crucial role in periodontitis, and it can be induced and stimulated by cytokines like IL-1β (Ozaki et al., 1996). Indeed, higher expression of MCP-1 was observed in the GCF and serum of patients with chronic periodontitis compared to healthy subjects (Emingil et al., 2004; Pradeep et al., 2009). We found that LpDNA effectively inhibited these cytokines and chemokine by regulating the MAPK and NF-κB signaling pathways. Additionally, LpDNA efficiently reduced TLR2 and TLR4 expression, implying that modulation of TLR expression may lead to inhibition of the inflammatory responses induced by P. gingivalis LPS through the suppression of MAPKs and NF-κB activation. These findings are in accordance with those of previous studies, which have shown that TLR2-JNK is the key signaling pathway activated during P. gingivalis LPS-induced cytokine production (Zhang et al., 2008). Moreover, it has been confirmed that P. gingivalis LPS-induced IL-1β production was inhibited by anti-TLR4 antibody in human gingival fibroblasts (Wang and Ohura, 2002).
Many studies have emphasized the role of MAPK and NF-κB in the development of periodontitis, particularly the importance of p38 MAPK signaling in periodontitis progression (Li et al., 2012). Treatment strategies to block signal transduction pathways, including MAPK and NF-κB pathways, have been developed to target periodontal diseases (Chopra et al., 2010; McIntyre et al., 2003). However, these inhibitors are associated with side effects due to the lack of specificity in modulating the host response (de Souza et al., 2012). Hence, novel therapeutic approaches that overcome these limitations represent promising alternatives for periodontal treatment. In this study, LpDNA was demonstrated to exert anti-inflammatory activities against P. gingivalis. These results highlight the potential of LpDNA as a novel postbiotic compound against oral pathogens, which may be implemented to effectively enhance oral health.