









Introduction
It has been well documented that collagen proteins play a pivotal role in tissue remodeling, wound healing and mainly toughness of meat due to an increase in the stability of cross-linking between the collagen molecules. Moreover, variations in collagen turnover may differ in different location of muscles in a same animal influence the meat tenderness (Archile-Contreras et al., 2010). For example, collagen concentration and collagen cross links are less in positional muscles such as longissimus dorsi (LD) muscle that leads to more tender and higher in locomotion muscles such as semimembranosus (SM) muscle leads to less tender (Dubost et al., 2013). Animal age can affect the collagen turn over then further affect the meat tenderness. Muscles from older animals have higher collagen and less tender than meat from muscles in younger animals due to less amount of collagen content and less cross-linking (Calkins et al., 2007; Jurie et al., 2005). Animal growth rate manipulates collagen solubility in meat but the studies of fibroblast growth rate on collagen characteristics have contradictory.
Fibroblasts as a predominant cells found in connective tissue are considered to be the primary source of the extracellular matrix components such as collagen. Fibroblasts provide tensile, compressive strength to organs and tissues, as well as the ability to adhere to other tissue types. The formation of new collagen fibers is essential for healthy and it is mainly present in skin, bones, tendons, cartilages and teeth. Collagen protein is widely used in food industry to improve the consistency, elasticity, and stability of foods in meat products (Taffin and Pluvinet, 2006).
Physical toughness of muscles and their variations between muscles are greatly associated with growth rate and extend of collagen type I and III. The collagen biosynthesis is very complex; about 34 genes are involved in collagen formation. The cytokine and growth factors genes are involved to synthesis of both type I and type III collagen through phosphorylation of the downstream mediators, Smad2/3in fibroblasts (Mun et al., 2014). Other than this, apoptotic and heat shock proteins/genes are also involved in collagen synthesis has been unclear (Alikhani et al., 2005). Even though, some genes are involved in collagen degradation in fibroblast cell. The synthesis of collagen has differed depends on muscle location. However, there are no reports on growth rate, collagen content, collagen synthesizing and degradation genes on positional muscles such as LD and locomotion muscles such as SM muscles fibroblast of Hanwoo.
Meat toughness is a function of collagen production through the generation of fibroblasts in muscle tissue. Taken this, the current study was designed to identify the phenotypic differences in the expressions of collagen synthesizing gene and collagen degrading genes between LD and SM muscles fibroblast of Hanwoo. Also we investigated that whether fibroblast growth rate could alter the production of collagen between the two fibroblast cells associated with meat tenderness.
Materials and Methods
The iScript TM cDNA synthesis kit (170-8891) and Sso FastTM Eva-Green® Supermix (172-5202) were procured from BIORAD (BIO-RAD Laboratories, Inc., Hercules, USA). Primers were obtained from Genotech (Korea). Dulbecco’s modified Eagle’s medium (DMEM) and fetal bovine serum were purchased from (Gibco®LifeTechnologies, USA). All the laboratory wares were purchased from Falcon Labware (BectonDickinson, USA). Collagen type I and Type III antibodies were purchased from abcam®#ab34710 & abcam®#ab 7778, Korea.
The entire work involving the use of animals was approved by an Institutional Animal Care and Use Committee (CBNU 2015-048 revised in 2015) at Chonbuk National University. Slaughter m. semimembranosus (SM) and m. longissimus dorsi (LD) muscles were excised from Hanwoo cattle (ca. 30 mon old) immediately after slaughter at a commercial abattoir (located in Jeonju, Korea). Immediately the sample is washed once with 70% ethanol and then quickly soaked in 1x PBS (phosphate buffered saline) containing 1% antibiotics (500 mL PBS in 800 mL sample beaker) to remove ethanol. Then soak the sample in DMEM containing 5% antibiotics and subsequently place it in an icebox before transferring to laboratory. After reaching the laboratory, sample is transferred to laminar flow hood. The muscle is excised, the top layers of tissue are trimmed and minced with fine sharp scissors [the samples are cut into small fragments (about 3 mm) using 60 mm petri dish with DMEM]. Five grams of minced muscle sample were incubated in DMEM (without antibiotics) + pronase (1 mg/mL, final concentration of enzyme is 0.1%) solution in a 15 mL sterile tube for 60 min with shaking (80 rpm) at 37°C. At the end of the digestion period, the muscle fragments were transferred to pronase solution to a 15 mL conical tube using a wide-bore Pasteur pipette. Subsequently, using a pipette with a narrow tip, the solution containing tissue fragments in pronase solution were pipetted until no clumps were visible. The suspension were collected and centrifuged at 400 g for 10 min. After centrifugation, the supernatant was discarded and the pellets were washed three times with 1x PBS and centrifuged, at the end of each step the supernatant is discarded. After the 3rd washing step, sediments were resuspended in DMEM and supplemented with 1% antibiotic/antimycotic solution and 10% FBS. Fibroblasts were then allowed to grow until about 95% confluence in 75 cm2 blank flasks in a humidified atmosphere at 37°C and 5% CO2. When the cells reached 95% confluence, cultures were split in a 1:10 ratio into new flasks with fresh media. At this ratio, confluent monolayers were obtained within 7 to 10 d after seeding. Selective detachment: the medium were aspirated from the flask, washed with PBS with mL of cold trypsin-EDTA (0.25%) and incubated for 2 min in the incubator and gently rap with open palm to release the fibroblasts and aspirate the trypsin-EDTA. This was added to a 15 mL centrifuge tube already containing 5 mL of DMEM+FBS solution and centrifuge. After centrifugation the pellets were collected and seeded into culture flasks.
To isolate myoblasts from the primary muscle cells, the cells were exposed to a magnetic cell sorting system (Auto MACS, Milteny Biotech, Germany). Particularly, when the cells reached 80% confluence, they were collected and resuspended in phosphate-buffered saline (1× PBS, Gibco) supplemented with 0.5% bovine serum albumin (BSA) and 2 mmol/L ethylenediaminetetraacetic acid (EDTA). After centrifugation (400 g for 5 min), the cell pellet was re-suspended in PBS containing 200 μL of primary rabbit IgG anti-fibroblast-specific-protein and then incubated with 20 μL of Anti- Rabbit IgG microbeads at 4°C for 30 min. Finally, cell suspension (approximately 107 cells in 2 mL PBS) was loaded into a magnetic cell sorting system to isolate fibroblast. After collection of fibroblasts from MACS separator, the purified fibroblasts could be cultured further for downstream experiments or stored in the liquid nitrogen. The isolated cells were cultured in growth medium, sub-cultured when they reached confluence (approximately 80%), and cells from the fourth passage were used for the present study.
Cell proliferation was measured with a Cell Counting Kit-8 (CCK-8, Sigma, Korea). LD and SM muscle fibroblast cells were seeded at a density of 5×103 cells/well in a 96-well plate for 24, 48, 72, 96, and 120 h, respectively. A colorimetric assay was used to create growth curves using the mean results from three independent experiments. The absorbance at 450 nm was measured with a plate reader according to manufacturer’s instructions.
1×105 cells were seeded into two T-75 culture flasks, one each for fibroblasts isolated from SM and LD muscle and cells were allowed to grow for 7 d. During these 7 d the medium in the flasks were not changed. At the end of 7th d, total collagen from the cellular layer was obtained with the addition of a protein extraction solution after rinsing the culture flasks with PBS. This solution consisted of a protease inhibitor cocktail in PBS. Then cellular layer were harvested by scraping at 4°C. Total collagen from the cellular layer was obtained by sonication of the cellular layer extract (3×15 s, with 30 s intervals, 20 Hz, 4°C). The sonicated samples were submitted to a collagen precipitation reaction with 25% saturated ammonium sulfate, for 24 h at 4°C, under constant agitation. Then collagens were isolated by centrifugation (40,000 g, 30 min, and 4°C). The discard the supernatant and the pellet were solubilized in 2 mL of 0.5 M acetic acid, then sample allowed to dialysis using dialysis membrane and stirred with 0.02 M Na2HPO4 for 48 h at 4°C. After this stirring time collect the pure collagen consisting of the aliquots of collagen from the cellular layer and the collagen was estimated by the following method.
The collagen content was estimated by Sirius red staining (Sigma-Aldrich Chemical Co., USA) according to the manufacturer’s instructions (Keira et al., 2004).
Cells grwon on glass coverslip in 12 well plates were cultured in DMEM. At the indication time, the cells were fixed for 10 min in 3% paraformaldehyde in PPS and then washed twice in PBS. Cells were permeabilized in 0.1% Triton X-100 in PBS for 10 min and then washed twice in PBS. Blockeing was performed in 3% BSA in PBS for 30 min and the cells were then incubated with collagen type I and collagen type III primary antibody (1:500 dilution, abcam®#ab34710 & abcam®#ab7778, Korea) overnight. After three wash in PBS, the cells were incubated with secondary fluorescein isothiocyanate (FITC) conjugated antibody for 1 h at room temprature and then washed three times in PBS. The coverslip were mounted on fluorescent mounting medium (Dako, France) and visualized under fluorescence microscope (Carl Zeiss, Germany).
After 80% of confluence cells were harvested and protein content was extracted using CelLytic M kit (Sigma) according to the manufacturer’s instructions. The protein concentration was determined using Bio-Rad protein assay kit (Bio-Rad). The sample containing 25 μg of protein were solubilized Laemmli buffer and separated by 6% acrylamide with 4% acrylamide stacking gels and then transferred onto the Hybond-P PVDF membrane (GE Healthcare, UK) for 60 min at 200 mA. Then the PVDF membranes were blocked using 5% skimmed milk powder in 0.5 M Tris-buffered saline, pH 7.4 with 0.05% Tween-20 (TBST) at room temperature for 2-4 h. Immunoblot were probed with collagen type I and collagen type III primary antibody (1:5000 dilution, abcam®#ab 34710 & abcam®#ab7778, Korea) overnight. After three time washing with TBST the membranes were probed with HRP-conjugated anti-rabbit secondary antibody (1:2000 dilution, abcam®# ab97051, Korea) for 60 min at room temperature. The membranes were then washed thrice with TBST for 10 mints each. Protein bands were visualized using enhanced Chemiluminescence assay kit (WesternBrightTM ECL, USA), the protein bands were scanned and quantified by Image J Software (version 1.44; Chonbuk National University). The relative density ratio of bands normalized to β-actin.
The cells were lysed using RNA extraction reagent kit (Tri-zol, Invitrogen) and total RNA was extracted according to the manufacturer’s protocol. cDNA was synthesized using 2 μg of total RNA by iScript TM cDNA Synthesis Kit from Bio-Rad. The quantitative Real-Time PCR (qRT-PCR) assays were performed using CFX96™ Real-Time PCR detection system (Bio-Rad). cDNA were amplified with each gene such as LOX, BMP-1, CYS, TRAP-1, TNF-α, NF-κB, caspase-7 caspase-9, HSP-70, HSP-90 and PPAR-γ and housekeeping gene GAPDH (Table 1). The reaction was carried out in 10 μl using SsoFast™ EvaGreen® Supermix (Bio-Rad) according to the manufacturer’s instructions. The statistical analysis of the qRT-PCR results was calculated by using the ΔCt value (Ct gene of interest-Ct reporter gene). Relative gene expression was obtained by ΔΔCt methods (ΔCt sample-ΔCt calibrator), with the use of unknown sample gene expression levels. The conversion between ΔΔCt and relative gene expression levels is as follows: Fold induction=2-ΔΔCt, where 2-ΔΔCt is relative gene expression (Livak et al., 2001).
Least square means and standard errors were estimated using a SPSS package (ver. 16.0). Replicates were included as a fixed effect and their significant levels were evaluated at p<0.05. The statistical analysis of the qRT-PCR results was calculated by using the ΔCt value (Ct gene of interest-Ct reporter gene). Relative gene expression was obtained by ΔΔCt methods (ΔCt sample-ΔCt calibrator), with the use of the sham-operated group as a calibrator for comparison of all unknowns ample gene expression levels. The conversion between ΔΔCt and relative gene expression levels is as follows: Fold induction=2-ΔΔCt, where 2-ΔΔCt is relative gene expression (Livak et al., 2001).
Results
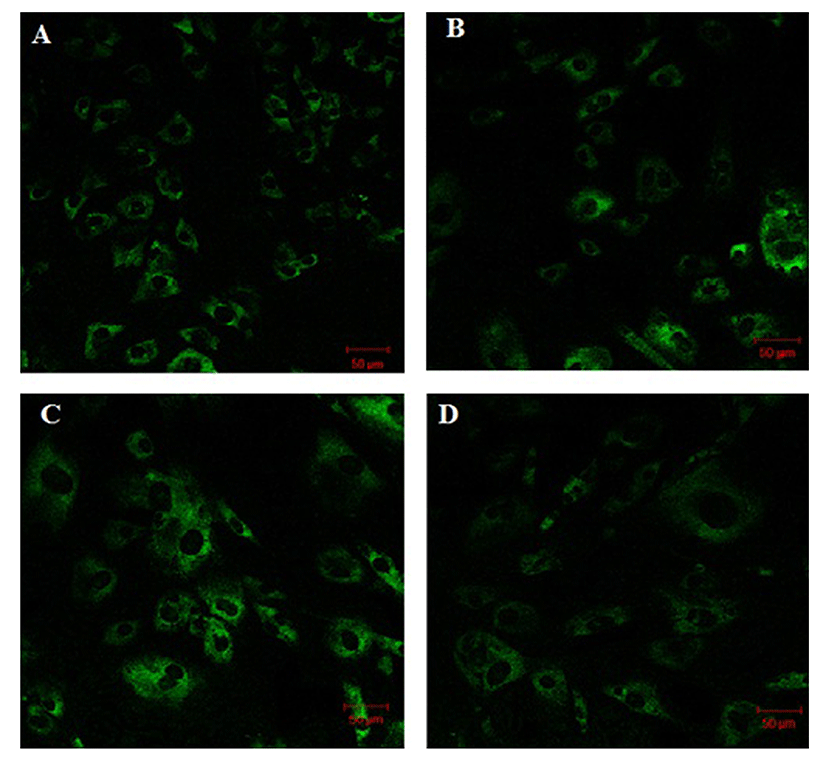
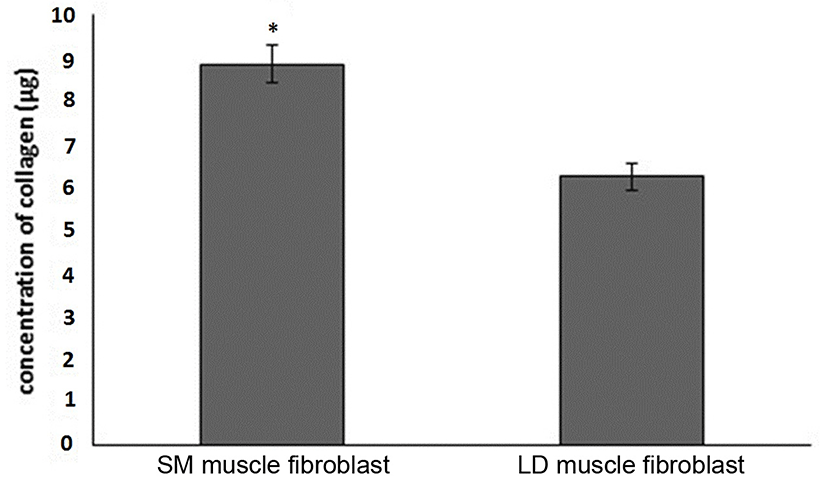
Fig. 1 shows that the cell proliferation rate of SM and LD muscle fibroblast cells was measured with CCK-8 assay kit. Curve was generated by reading the absorbance value at different time point (for 5 d). The fibroblasts of SM muscle cells were significantly higher cell proliferation than LD fibroblast cell. Fig. 2A, 2B, 2C and 2D shows the immunohistochemistry of collagen type I and type III in two different fibroblast cells derived from SM muscle and LD muscle. SM and LD muscle fibroblast containing both collagen types (I and III) although increased fluorescence of collagen type I and type III appears stronger in SM than LD muscle fibroblast cells. The total collagen content in each cultured fibroblast cells was measured. Fig. 3 shows the percentage of total collagen production was significantly higher in SM muscle fibroblast (8.85 g) compared to LD muscle fibroblast (6.25 g). Fig. 4A depicts the western blot analysis of collagen type I and type III in SM and LD muscle fibroblast cells. The relative density values of the bands were normalized with β-actin and expressed as a ratio (Fig. 4B). Our data reveals that collagen type I and type III protein were significantly higher in SM muscle fibroblast than LD muscle fibroblast. This is confirming that the collagen type I and type III protein highly present in SM muscle fibroblast than LD muscle fibroblast.


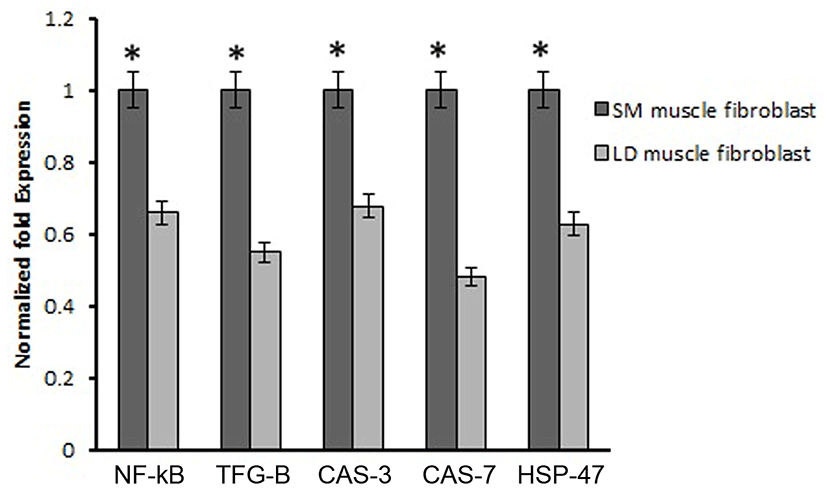
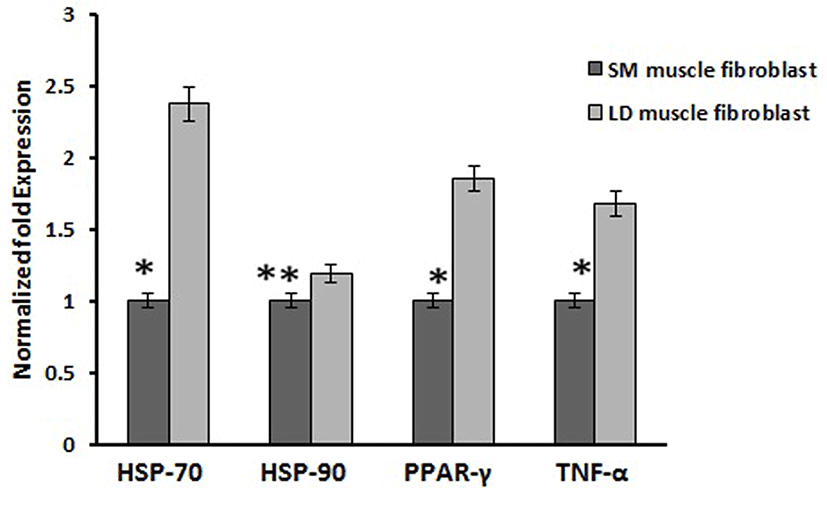
Based on this, we analyzed the mRNA expression of collagen synthesizing genes and degrading genes on cultured fibroblast from SM and LD muscle fibroblast cells of Hanwoo. The mRNA expression of collagen synthesizing genes such as LOX, BMP-1, CYS and TLP-1 were significantly decreased in LD fibroblast cells compared to SM muscle fibroblast cells. The decreasing levels of the LOX, BMP-1, CYS and TLP-1 in LD muscle fibroblast were 79.2, 56.8, 58.9 and 29.8%, respectively compared to SM muscle fibroblast (Fig. 5). The mRNA levels of NF-κB, TGF-β, CAS-3, CAS-7 and HSP-47 were significantly (p<0.05) higher (67.9, 55.0, 67.8, 48.2 and 62.8%, respectively) in cultured SM muscle fibroblast cells compared to LD muscle fibroblast cells (Fig. 6). We next examined the effects of collagen degrading genes such as HSP-70, HSP-90 PPAR-γ, and TNF-α on cultured fibroblast cells of SM and LD muscle. The levels of HSP-70, HSP-90, PPAR-γ, and TNF-α were increased 57.94, 15.89, 46.03 and 40.47%, respectively in LD muscle fibroblast when compared to SM muscle fibroblast cells (Fig. 7).

Discussion
Fibroblast derived from LM had least proliferative capacity than semitendinosus (STN), and sternomandibularis (SMD) muscle fibroblast cells. The in vitro primary culture of fibroblast cells from different muscle exhibit dissimilarities in growth behavior and cell expression (Dubost et al., 2013). Our study shows that the fibroblast cells from SM muscle having higher growth rate than LD muscle fibroblast which suggested that locations of muscle could affect the fibroblast growth capacity are responsible for collagen synthesis. Due to increased growth rate and collagen synthesis can affect the meat tenderness than lesser growth rate of fibroblast. Furthermore, fibroblast cells may be grow faster leads to produce more collagen and more toughness in locomotion muscles than position muscles not only muscle also breeds and sex. It is evident from Fig. 3 that the collagen concentration was higher in the fibroblast of SM muscle than fibroblast of LD muscle fibroblast may be according to growth rate. We speculate that same mechanism might be occurring in vivo condition. For example, LD and Gluteus medius (GM) muscle contained lowest collagen content and more tenderness than SM, biceps femoris (BF) and semitendinosus (ST) muscles (Stolowski et al., 2006). Hence, the muscles of BF and ST might have higher fibroblast proliferation to make higher production of collagen than GM and LD muscle.
The proportion of type I and type II collagen in intramuscular connective tissue is only one several connective tissue characteristics that can be (imperfectly) correlated to general patterns of tenderness variations between muscles. For example, collagen type I or ratio of type I and type III are main indicator of tenderness in different muscle type, breeds, and sex. Tenderness in meat is associated with the amount of collagen in muscle and amount of cross-linked collagen, which increases as growth slows when animal matures. Moreover, connective tissue is present in all muscles in the form of collagen especially collagen type III, So that reason collagen type III is main indicator of tenderness in the form of connective tissue. For example, Criolla Argentina breed had a less tenderness which containing more percentage of collagen type I than Aberdeen Angus breed. Similarly, GM muscle containing more collagen type I and less tenderness than LD muscle (Blanco et al., 2010). It is believed in our study (Fig. 4A and 4B) that the SM muscle fibroblast occurring more collagen type I and III than LD muscle fibroblast, which cause inconsistency in meat tenderness. Additionally, many researchers have investigated that changes in tenderness based on types of collagen mainly I and III and collagen stability (Tatum, 2011). However, here we show for first time that the growth rate of SM fibroblast cells was coordinate with expressions of collagen (type I and III) in fibroblast cells from SM and LD muscle related to differences in meat tenderness.
The biosynthetic pathway of collagen production is a very complex (Mun et al., 2014). Specific collagens are encoded by specific genes; COL1A1 an COL1A2 for type I collagen and COL3A1 for type III collagen, are may involve in the regulation of collagen synthesis in animal (Gonzalez et al., 2014). Differences in LOX, BMP-1 and CYS expression between the muscles have been reported. Although, first time we elucidated that mRNA expression of LOX, BMP-1 and CYS in two different fibroblast cells derived from Hanwoo muscle. LOX is secreted in fibroblast which has responsible for collagen formation synthesis via oxidizing specific peptidyl lysine residues leading to stabilization of the collagen (Sambasivarao, 2013). On the other hand, LOX would be regulated by some cytokines such as TGF-β, IL-6, IL-12 and NF-κB, these cytokines are also involved in collagen formation (Verrecchia and Mauviel, 2004). An in vitro model describes that TGF-β and C-proteinase (encoded by BMP-1 gene) can induce the LOX gene in human trabecular meshwork cells. Thus, the mRNA expression of LOX could be controlled by TGF-β and C-proteinase (Uzel et al., 2001). Likewise, BMP-1 has been important for collagen-synthesis and collagen remodeling in fibroblasts. These are consistent with earlier reports, suggesting that the activity of LOX and BMP-1 may directly stimulate the collagen synthesis in dermal fibroblasts via the activation TGF-β (Kessler et al., 1996). In addition, reducing activation of CYS leads to enhance collagen degradation by inhibited the lysosomal cysteine proteases like Cathepsin B (Namboodiripad, 2014). Also, knock down of cysC−/− mice had shown lowest collagen content compared to normal mice (Chapman et al., 1997). Based on our finding we proposed that LOX, BMP-1 and CYS might be played a crucial role in collagen synthesis in SM muscle fibroblast than LD muscle fibroblast of Hanwoo. From our results we speculate that the expression of LOX, BMP-1 and CYS might influenced by fibroblast growth rate associated with location of fibroblast in muscle. Similarly, increased LOX and BMP-1 enzyme activity can lead to excessive accumulation of collagen in cardiac fibroblast this may be related to in vivo (Kagan, 2000). For example, the expressions of LOX, BMP-1 and CYS were greater in Brangus cattle than Angus than Brahman cattle because of Brangus containing more collagen than others. In our study, the fibroblasts derived from SM muscle shows higher expression of LOX than LD (Fig. 4). This is indicating that different rate of collagen turn over in different muscle may facilitate the LOX activity through fibroblast. Therefore, higher levels of LOX may be associated with increased synthesis of collagen which may affect the meat tenderness (Lindahl et al., 2002).
Moreover, the activation of TGF-β involved in the production of collagen by inhibiting the synthesis of extracellular proteases which can be degraded the collagen synthesis. Furthermore, the expression of type I collagen is dependents on TGF-β activity in fibroblasts and smooth muscle cells (Kim et al., 2002). In addition, TGF-β as a positive mediator of collagen accumulation in normal kidney fibroblast cells and stimulates the total collagen content in chicken embryo fibroblast. Hence, TGF-β upregulates the expression of collagen, and decreases the collagen degradation by suppressing the proteolytic degradation of the ECM components (Ziegelhoffer-mihalovi et al., 2003). Similarly, TLP-1, a newly discovered cytoplasmic protein, plays an important role in the synthesis of collagen type I & III in human dermal fibroblasts by regulating Smad2, Smad3 in TGF-β pathway. Knockdown of TLP-1 significantly reduced the synthesis of collagen type I and III and also reduced cell contraction in hypertrophic scar fibroblasts, compared to control groups (Wang et al., 2013). Based on those support, our current study suggested that the expression of TGF-β and TLP-1 genes are play a vital role in production of collagen formation in SM than LD muscle fibroblast cells (Fig. 5).
NF-κB and HSP-47 are an another important principal mediator of collagen formation through the activation of MMP-9 in cardiac fibroblast. As previous reports showed that the knockdown of NF-κB and HSP-47 in fibroblast cells results reduced collagen synthesis (Nakai et al., 1992; Shioshita et al., 2000) Thus, the activation of NF-κB and HSP-47 could directly promote collagen formation in fibroblast cells through IL-6. In addition, NF-kB regulates the collagen type I in fibroblast cells (Chen et al., 2012; Kalayarasan et al., 2008). Similarly, changes in the expression of HSP-47 in cells are associated with changes in the levels of collagen (Enjalber et al., 2006). In the present study has demonstrated that the mRNA expression of NF-κB and HSP-47 was highly contributed to collagen synthesis in SM muscle fibroblast than LD muscle fibroblast cells (Fig. 6). Thus, NF-κB and HSP-47 might be stimulating the collagen formation depending depends on muscle types and fibroblast.
Caspase-3 is another important biological process which has been associated with regulation of collagen formation in fibroblast (Ruiz-Ortega et al., 2006). Our result is consistent with the previous reports showing, that the collagen production has enhanced by the activation of caspase including caspase-3 and caspase-7. Conversely, HSP-70, HSP-90 and PPAR-γ are not master gene for collagen synthesis which has not associated with collagen synthesis (Peng et al., 2011). Those genes are act as negative role in the synthesis of collagen. Likewise, activation of PPAR-γ may attenuate the connective tissue target genes leads to inhibition of collagen synthesis by blocking of TGF-β. In addition, increased the protein expression of PPAR-γ directly inhibits the procollagen type I in human hepatic stellate cells (Sun et al., 2006). In the same way, Ajulemic acid suppresses the synthesis of collagen via activation of PPAR-γ. Increase in expression of PPAR-γ might be involved in the collagen degradation process by blocking of TGF-β but the mechanism by which PPAR modulates the collagen been unclear (Galli et al., 2002). However, in our study the mRNA expression of HSP-70, HSP-90 and PPAR-γ were significantly down regulated in higher collagen containing SM fibroblast than LD muscle fibroblast (Fig. 7).
Conclusion
Fibroblast cells isolated from SM and LD muscle of Hanwoo in the same animal shows variation in proliferative rate and collagen content. The differences in the expression of collagen in muscle due to the variation in growth rate of fibroblast cells had significant effects on meat tenderness and toughness. The two different muscle fibroblast cells have responded differently each other in collagen synthesized genes such as LOX, BMP-1, CYS and cytokines namely NK-κB, TGF-β as well as apoptotic signal like caspase-7, caspase-9 and HSP-47. The variations in those mRNA expression in each muscle is depends upon the fibroblast’s growth rate and location of muscle. Moreover, the reduced mRNA expressions of HSP-70, HSP-90, PPAR-γ and TNF-α are more responsible for degradation of collagen in LD muscle fibroblast than SM muscle fibroblast. These findings indicate that the collagen synthesis in fibroblast has been regulated by some genes are differed due to variations in fibroblast growth rate in muscle to muscle leads to vary the meat tenderness. The increased collagen synthesized genes are the possible mechanism would be at genetic level to development of toughness in meat. Also, locomotion muscles having higher fibroblast growth rate which can synthesis the more collagen than positional muscle fibroblast. These same mechanisms from in vitro findings may be related to in vivo animal associated with differences in meat tenderness.